


Very, very few chronic fatigue syndrome studies have emerged from Germany, but the last two have been good ones. The one before this was the first to find evidence of EBV activation in ME/CFS, in years.
This one – a model exploring mitochondrial dynamics – may help explain what’s causing the post-exertional problems, published in the Biophysical Chemistry journal, this study extended a well-known metabolic model explaining what happens to the mitochondria in the skeletal muscles during exercise. The authors enhanced it by adding some processes to it (lactate accumulations / purine degradation) known to occur in the mitochondria.
In silico analysis of exercise intolerance in myalgic encephalomyelitis/chronic fatigue syndrome. Nicor Lengert, Barbara Drossel Institute for Condensed Matter Physics, Technische Universität. Biophysical Chemistry 202 (2015) 21–31
The Study
The model covers two parts of the inner mitochondria – the cytosolic space and the mitochrondrial matrix. It does not cover interactions between the mitochondrial membrane and the rest of the mitochondria.

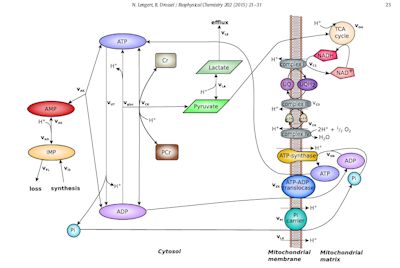
The model was rather….complex.
The production of the model was sparked by a number of findings (reduced ATP production and peak oxygen uptake) suggesting problems with aerobic (oxygen related) energy production, were present in chronic fatigue syndrome. Findings of increased acidification, reduced anaerobic threshold and prolonged pH recovery times suggested that anaerobic respiration – a less efficient and more toxic form of energy production – was attempting to compensate for a broken aerobic energy production system.
They noted that several factors that could affect mitochondrial activity. They included possible “mitochondrial deletions”, Epstein-Barr virus induced alterations of mitochondrial gene transcription, pro-inflammatory cytokines and increased levels of oxidative stress.
They ran two iterations of the model; one representing the reactions in the mitochondria occurring in healthy controls, and one representing a person with probably severe ME/CFS, for whom mitochondrial capacity was reduced by about a third. Then they examined what happened during three exercise scenarios: 30 seconds of intensive exercise, an hour of moderate exercise and an hour of moderate exercise spread across two days. Then they looked to see if the models fit what the studies suggest is happening in ME/CFS.
It turns out that they did.
Results
ATP reaches critically low concentrations during high intensity exercise in CFS simulations and the acidification in muscle tissue increases compared to control simulations. Authors
Several studies suggest that the rates of ATP production/oxidative phosphorylation (mitochondrial capacity) are about 65% of normal in ME/CFS. This model suggests reduced mitochondrial capacity could be causing the ATP problems and the increased acidosis and lactate accumulations found in several studies. (The increased acidosis is the problem, lactate is not. Lactate is produced to protect the cell from acidification.)

The model suggested that the muscles cells in ME/CFS poop out quickly during exercise and then cannabalize themselves in an attempt to keep going.
Healthy people are able to maintain an ATP level during exercise that protects their mitochondria. The models suggested, however, that the minimum ATP levels maintained in ME/CFS patients during exercise, may be significantly lower – low enough perhaps to induce cell death. This finding was buttressed by one of Sarah Myhill’s studies, which found greatly increased levels of a factor (cell-free DNA) that’s associated with increased cell death.
When the mitochondrial capacity of a cell is exhausted; i.e. when ATP demand outstrips supply the cell copes with this by digging into its reserves. Converting two ADP molecules to one ATP and one AMP molecule, frees up some energy (ATP), but does two negative things as well. First, it reduces the total adenine pool in the cell (adenine triphospate (ATP) and diphosphate (ADP)) and second it increases levels of inosine, hypoxanthine and finally uric acid.
The model suggested that the healthy controls in the model were able to exercise without purine nucleotide loss while the people with reduced mitochondrial dysfunction suffered from significant losses of purine nucleotides.
Acidosis Plays Key Role
Anaerobic respiration greatly increases the rate of acidosis. Acidification is produced by the breakdown (hydrolysis) of ATP and is related, if I have it right, to increased rates of cell damage and subsequent purine nucleotide loss. The model suggested that the increased acidosis in the “ME/CFS patients” increases lactate accumulations and lactate efflux from the cell by 10-15%. The situation in ME/CFS might be worse, however, than the model with its forty percent reduction in mitochondrial production suggested. The rates of acidosis and lactate accumulations found in ME/CFS studies were significantly higher than those produced by the model.
Prolonged Recovery Periods
..the model…. demonstrates that long moderate exercises are more exhaustive than short intensive exercises contrary to the results for healthy controls. Authors
The reduction in the adenine pool means the cell will need time in the post-exercise period to get back to normal. After prolonged bouts of intense exercise even professional athletes need 72 hours to replenish the adenine pools in their muscle cells. But what about short periods of exercise? The model predicted it would take 3-5 times longer for the ATP levels in the muscles of ME/CFS patients to return to normal after exercise than for healthy controls. The model also predicted that short (30 seconds), intense exercise periods would be easier for ME/CFS patients to recover from.

The resulting depletion results in long recovery periods.
It predicted it would take 49 hours for ATP levels in the muscles to return to normal after a longer (30 minutes) but more moderate period of exercise. It would take 32 hours for the ATP levels in the muscles of ME/CFS patients to return to normal after short but intense (30 secs.) periods of exercise.
The model’s prediction that mitochondrial depletion results in more difficulty with longer bouts of moderate exercise than with shorter bouts of intense exercise was opposite to that found in the controls. The altered scenario fits well with recommendations by exercise physiologists for ME/CFS suggesting that exercise periods be short and interspersed with rest periods.
The findings provide another possible reason (ATP depletion) for the post-exertional malaise seen in ME/CFS. It could also explain why some people with ME/CFS cannot reproduce their levels of energy on the second day of a bicycle exercise test.
The model did not account for the possibility of increased cell death during exercise – something the researchers thought likely – which would extend recovery times further. Other factors, such as sympathetic nervous system and immune dysregulation and increased oxidative stress not included in the model may also come into play.
Two Subsets?
Studies of mitochondrial respiration in the neutrophils in ME/CFS suggested two groups of mitochondrial deficient ME/CFS patients may be present. One group compensates for the mitochondrial deficiency by upregulating glycolysis and the other by increasing purine nucleotide degradation. This model in this paper suggests both are possible.
Treatment
The authors suggested that D-Ribose could help in the short term but worried about its “rapid glycation of proteins” – something they said was associated with some neurodegenerative diseases. They suggested measuring mitochondrial respiration co-factors (ubiquinol (CoQ10), NAD, L-carnitine and others) and supporting it with supplements.
Wrap Up
The new model produced in this study extends a less sophisticated model of mitochondrial metabolism. That fact that it was produced in response to findings in ME/CFS suggests how unusual the findings in ME/CFS exercise studies are. It should be emphasized that this is a model, and therefore does not necessarily demonstrate what’s happening in ME/CFS.
The model indicates that reduced mitochondrial production could, however, help explain the exercise findings, and post exertional malaise studies indicate it is present in ME/CFS. It buttresses the idea, emerging more strongly in ME/CFS in recent years, that the mitochondria may play an important role.
It did this by showing how, when put under load, reduced mitochondrial capacity causes the cells to reach into their reserves to make energy. This works in the short term, but in the longer term depletes the cells of vital energy making factors. This long term depletion – lasting for at least a day, but possibly much longer – could help explain the post-exertional malaise and difficulty reproducing energy production, in the second day of a two day exercise test.
It indicated that shorter periods of exercise – even if they are more intense – should be easier to recover from, than longer periods of more moderate exercise. Because the model failed to predict the very high levels of lactate and acid production found in some ME/CFS studies, it may be understating the depth of the mitochondrial depletion found. It does not say anything about what is causing the possible depletion of mitochondrial capacity.
The authors suggested the appropriate supplementation (CoQ10, NAD, L-carnitine) may be helpful.



My muscles began to ache and I had fatigue spells after starting a regimen of 80mg per day of simvastatin, then 40mg atorvastatin. On stopping the statins the symptoms have continued and possibly worsened over the last 9 years. Since statins are known to deplete CoQ10, dolichols etc, could this be the cause of my FM/CFS?
I believe there is a typo. “adenine triphospate”, solid be adenosine…
Thank you for this very interesting and informative article.
“But what would short periods of exercise to please with reduce exercise capacity?”
Is this a typo? I don’t understand.
(Note: Please feel free to delete this comment. With brain fog submitting a comment is easier than trying to find your email address and send a message.)
I need to reread and try to better absorb this info…but even with what I’ve gleaned so far (hopefully without misunderstanding), I feel like it might support my own feeling that having homozygous AMPD1 (myoadenylate deaminase) deficiency mutations could relate to my own CFS. (I’m not officially diagnosed with CFS, but feel sure that I could be if I pursued it.). I’m also curious on EBV reactivations and what they look like in the lab results…I can’t tell from the weird Labcorp ranges whether my IGG is unusually high or instead is just the normal “you had mono sometime” range.
You can request a “PCR” of your blood for EBV. This test looks for live virus, so you either show an active infection or you don’t. It’s black and white, rather than the grey of an EBV titers test.
I know what you mean !! The doc that school my cfs said it was likely caused by multiple bouts of mono and finally EBV. My present doc says its useless to test for this since there are no treatments for it. He seldom does blood test at all. But he is one of the few docs that would accept me as a patient since I have fms/cfs. Others turned me down flat.
What I don’t get about d-ribose is that it’s just a simple five carbon sugar. How can it possibly make it through the stomach without all getting digested?
I vaguely remember someone saying/reading EPO may be useful in this. Would this help recovery of muscles and other things described above? Would this also fit with Fluge Mella etc by increasing mature red blood cells via just having more red blood cells with EPO? Just a layman sufferer 🙁
i wonder too with b12 supplementation if the pro-blood cell effects could help in this case.
Are you referring to vitamin b12 that we take everyday ?
Hi Brenda,
The methylcobalamine form of B12 is the most potent as it crosses the blood brain barrier, however you’d have to take a lot of pills to equal what you would get by injection. Maybe talk to your doctor about getting injections. I find they help but don’t last more than a week. It’s also fairly expensive, in Canada it’s about $40 for 2 injections. I take mine every 2-4 weeks.
I remember hearing/reading EPO might be helpful in this. Could it help with recovery of muscles etc as described above? Could it also help with Fluge Mella etc theory on not enough mature red blood cells by just bumping up the numbers of them as a whole. Just a layman suffer 🙁
OR…get the EBV out? 😉
This just makes so much sense to me. I can almost feel this process happen ing (or not happening !) in my thighs. I would recommend dr Myhills mito testing. I’ve had it done twice and it gives me a sense of where I am in my recovery
the paragraph titled ‘recovery reversal’…when stating that long bouts of 4.5hrs vs short bouts 10.3 hrs…did you mean 10.3 mins.???
The times are recovery times not exercise times. So if healthy people do moderate exercise for 30 minutes they recover in 4.5hrs, if they do short intense exercise for 30 seconds they take 10.3hrs to recover. CFS/ME patients not only take much longer to recover (more than 24hrs) they recover quicker from the short intense exercise (30 seconds intense exercise, 32 hours to recover) than the moderate long duration exercise (30 min exercise, 49 hours to recover).
Maybe there is another part to this. An enzyme, lactate dehydrogenase inter converts lactate and pyuvate. I have been tested for LDH and am consistently 60 to 70 per cent of normal values. ( about 5 evaluations) Lack of LDH is considered a glycogen storage disorder, type XI.
Found one study : Glycolysis abnormalities in fibromyalgia by Eisinger J et al. J. Am Coll Nutition. 1994 Apr; 13(2): 144-8. They concluded that ATP and LDH muscular enzymes were decreased in Fibro , and that glycolysis was impaired in fibromyalgia.
Also, mature red blood cells have no mitochondria and depend on glycolysis for energy production. Any biochemists out there who can put this together to make sense???
My LDH has also been well below the reference range every time it is tested!
Now I am curious if this is a common finding?
I think kreb cycle intermediaries such as ldh are commonly off in fibro. phenyl butyrate can lower lactate- and butteris high in butyrate. Oxidized atp, SAM E and mito q might help, as well as nadh or other kreb cycle intermediaries. My own personal belief is that it will take some time with the right treatment before the epigenetics of fibro change-and the change needs to be dramatic, of course. This might require radical change in diet/lifestyle.
Hmmmm. Interesting.
I don’t have fibro, and pain is not a major part of my illness.
Before ME/CFS I used to exercise a lot including cycling, swimming and hiking definitely aerobic of at least one hour duration. I had taken a short kinesiology course a few years before and remember learning the physiology of exercise, how the mitochondria function, Vo2, lactic acid build-up, recovery etc.
When I became sick I could no longer exercise anywhere close to my usual capacity. I tried to explain to my doctors/specialists that it felt like my cells were energy depleted and the build-up of lactate was quicker, worse and longer lasting. Now I’m not a scientist so could never really explain or understand exactly what was going on or why. I’m just patting myself on the back for realizing I may have been on to a theory of what was happening to my body long before any doctor would listen, I feel a little vindicated, sounds petty but when you go for years and years and feeling nobody is listening, well, I’ll take anything positive.
ME/FM is complicated and I’ve come to realize there are many theories leading to it’s etiology and it may be awhile yet before the mystery unravels, but reading Cort’s blog gives me hope that one day there’ll be an ah-ha moment in a future trial. Gotta keep looking forward. I’m sounding quite positive today, must be the nice warm sunny day we’re having, plus I slept well-yay!
Really insightful. Before brcoming sick I weight lifted 3 days a week and I would get lactic acids exactly 24 hours post workout and it would taper off over a period of 1-2 days. Now, even in remittance months, when I’m doing about 20% of capacity my lactic acid build up hits earlier and lasts 5-7 days.
I also have decided (if I’m ever well enough to make it to the gym again) to ditch cardio completely and just weight lift. Based on this article I will attempt max weight for 30 second bursts. I wish I knew how long to wait between each burst!
Danielle,
If you look up Dr. Mark Van Ness “Exercise and ME/CFS” you can watch a video he presented at the Bristol Watershed. If I remember correctly, he recommended 30 seconds of exercise followed by 2 minutes of rest, with the whole exercise period not to exceed 20 minutes. He also said something about an activity-to-rest ratio of 1:3. He suggested doing light weights, step ups, or stretches during this exercise period.
One other thing he mentioned regarding aerobic exercise was “why would you try to train a broken (aerobic) system”. I found that disturbing and depressing, probably because it seems to be true for me.
Dr. Van Ness is with the Workwell Foundation.
Kathy
Could mitochondrial depletion be expressed via high amonia levels in the blood?
I think mitochondria can lead to nitrosative stress and accumulation of ammonia-and ammonia is as toxic as lactic acid. Phenylbutyrate can lower ammonia levels and lactic acid-and butter is high in butyrate.
I have asked this question before and I do hope I am in the right place to ask this.
I have long term cfs/me/fibro and I do believe lyme is in the mix. I am very knowledgeable having this as long as I have. Unfortunely, I wish the docs knew at least half of what I know. This is my issue. I need a doctor or practicioner that is knowledgeable and if possible specializes in cfs/me/lyme literate. Right now I have no doctor as he no longer takes my insurance. I feel I need to have some specilalized testing done as I have noticed some no so good changes in my health. I am open to a researcher,biochemist. An M.D. would be my first choice. I live in Ma. I am close to Boston and surrounding areas.
I would appreciate it,if any-one out there knows of any docs treating this in the Ma. area.
Thank-you so much,
I do appreciate any info you can give me.
Sincerely,
Lynn Calderaro