


Effective pain management is a moral imperative – one we are not meeting. An IOM report
Pain management is dominated by the use of opiates but arguably it shouldn’t be. Producing a sensation of pain is a complex operation. From the pain signals produced at the site of an injury, to the peripheral nerves that transmit those signals, to the dorsal root ganglia that filters them, to the spinal cord and then up to the brain, the production of the pain involves many factors – each of which could conceivably be targeted to reduce pain.

Turning off chronic pain was the subject of the webinar
This blog uses a Pain Research Forum webinar “Targeting Unusual Voltage-Gated Sodium Currents for Pain Therapy” to check out work that’s targeting one of the first players on the scene – the sodium channels in the sensory nerves that start the process of producing pain in the body.
Ted Cummings, a pain researcher, started off the webinar by noting that the central nervous system plays a large role in the production of chronic pain. Turning down the pain signal coming from the body could enable the central nervous system to take its foot off the pain pedal.
Cummings wants to turn off the most basic aspect of pain production – the increased electrical activity in the sensory nerves which causes them to shoot pain signals to the brain.

That means focusing on the”voltage-gated sodium channels” that litter pain-producing neurons. When these sodium channels open, sodium floods into the cell causing it to generate an electrical charge – which sends a pain signal zinging up the nerves to the brain.
Cummings said his lab has studied these sodium channels in “exquisite detail.” Nine different types of sodium channels exist. If you’re interested in targets for pain then three types of sodium channels (Nav 1.7, 1.8, 1.9) jump out.
Nav 1.7 sodium channels have been the best studied, but Nav 1.8 and 1.9 sodium channels may provide the best target. The goal in drug development is to find a target that only applies to pain. The Nav 1.8 and 1.9 sodium channels mostly meet this criterion both mostly found only in the periphery in pain-sensing neurons. That means that knocking them down won’t make other parts of your body go kablooey. Genetically they are very distinctive. That’s a good sign – it means that a drug that affects them probably won’t affect some other part of the body.

The wallop the Desert Bark Scorpion delivers feeds through sodium channels
How much pain are these little ion channels responsible for? Consider the case of the Arizona Bark Scorpion which produces one of the ten most painful stings known to man. The Arizona Bark Scorpion’s sting has been described as having a burning cigarette ground into your arm and then a nail pounded through it.
Not so for the desert mouse. The bark scorpion’s sting can kill lab mice but has no effect on the desert mouse that eats scorpions like candy. Studies indicate that a scorpion’s bite turns or excites the pain nerves in laboratory mice but actually deadens them in the desert mouse. The more the desert mouse is stung – the more impervious to pain it gets. The scorpion’s sting is an analgesic – an opiate – for desert mice. The ion channels that it either turned on or shut down, depending on which mouse was stung, were the Nav 1.8 channels.
Nav 1.7 ion channels also clearly associated with pain. They’re found primarily in nodules (the dorsal root ganglia (DRG)) found outside the spinal cord that filter sensory signals from the body. (The DRG are also commonly colonized by herpesviruses). Mutations in these ion channel genes allow the ion channels to open more easily – allowing them to translate small amounts of stimuli such as touch into pain. Sound familiar? In 2012, Dr. Martinez-Lavin found that a mutation in Nav. 1.7 genes was associated with severe fibromyalgia.
(Martinez-Lavin believes that infections such as herpesvirus infections and/or trauma cause the sympathetic nerves in the DRG to “sprout” leaving ion channels more sensitive. Since Nav 1.7 is found on both sympathetic nervous system neurons and other DRG neurons, Martinez-Lavin postulates that these gene mutations cause both the SNS and pain producing neurons to become hypersensitive to stimuli – hence Martinez-Lavin’s postulate that stress causes distress or pain in fibromyalgia.
Resurgent Electrical Impulses – Resurgent Pain Production
These sodium channels usually become activated and then quickly become inactivated and enter into a quiescent phase. A close study of them however, indicated that in chronic pain these ion channels “resurge” or open up during the inactive period and start dumping sodium back into the cell. This inability to fully turn off the electrical signal produces an always “on” or hyper-excitable pain response.
Cumming’s believes these resurgent currents are intimately involved in producing chronic pain. His and other labs indicate they have found that a wide variety of pain producing factors including inflammatory mediators and many biological toxins (wasp, scorpion, sea anemone toxins), induce these sodium ion channels to produce resurgent electrical currents; i.e. ongoing pain.
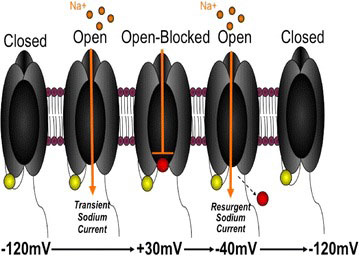
Resurgent electrical currents appear to be behind chronic pain states.
The big question is whether a drug can be developed to turn this resurgent electrical off. If it could, the chronic pain production process could conceivably be shut down at the source. Cummins thought it was possible.
In a paper produced earlier this year, Cummin’s reported that he’d found a particular genetic target called NavB4 which is able to stop some ion channels from resurging. When they knocked out NavB4 subunit in the lab all the pain issues (allodynia, resurgent electrical currents, Navb4 and spontaneous pain nerve fiber activation) resolved. Now the question becomes how to produce a drug that can activate that genetic unit.
The Webinar
Current Drugs
In what would be a recurring theme it was clear that some drugs already on the market can stop this resurgence of electrical activity but the participants felt much more study was needed to identify which drugs in which patients were able to achieve this. All the drugs identified thus far appear to stop the resurgence in different ways.
Dr. Christine Sang noted that several anti-epileptic drugs effected sodium channels while cannabidiol (from Cannabis and lidocaine can jam up the sodium channels and stop them from activating again.)
In the short term finding old drugs that block sodium channels is a priority
A review of the recent literature found that several drugs have been identified.
- A congener of Sumatriptan, a migraine drug knocked down Nav1.7 ion channel activity
- The anti-arrhythmic drug Mexiletine knocked down Nav1.7 activity.
- Rufinamide stabilized sodium channel activity in mice with neuropathic pain.
- Lacosamide, a sodium channel blocker, is being tested in patients with a kind of small fiber neuropathy associated with sodium channel problems.
- A synthetic cannabinoid called ajulemic acid derived from THC (but which does not produce a high) inhibited the activity of all forms (Nav1.2-1.8) of sodium channels tested.
- An ongoing study will determine whether carbamazepine improves brain functioning in erythromelalgia patients with sodium channel problems. Carbamazepine is currently used mostly in epilepsy and neuropathic pain.
New Drugs
Genentech scientist David Hackos noted that Genentech is working on a new class of small molecules that will lock sodium ion channels in their quiescent phase period which occurs directly after activation. This is a very useful approach because it turns off ion channels when they’re active and causing problems, but allows them to become active again when appropriate. That obviates a concern that sodium channel blocking drugs could turn off pain signals entirely – causing patients to injure themselves unknowingly.
Genentech’s focus is mostly on a well-studied and validated ion channel called Nav 1.7. Hackos noted that multiple companies were working on Nav1.7 ion channel blockers. That comment was echoed by another member of the panel, Christine Sang, who stated that Pfizer was working on a Nav1.7 blocker.
Hacko’s said Genentech has developed an intricate knowledge of how different compounds effect ion binding channels. In fact, Genentech, Pfizer, and others appear to have developed a knowledge of a specific binding site on sodium channels they believe they can use to develop effective drugs. Hackos stated that he hoped Genentech would be able to use this knowledge to develop “really remarkable” pain drugs in the future.
Another panel member noted that several compounds coming online had limited side effects. He suggested that several sodium channel blockers or sodium channel blockers in combination with other drugs might be necessary to extirpate pain.
Some New Drugs
- Pfizer has completed phase II trials in erythromelalgia (EM), wisdom tooth pain and peripheral neuropathy in diabetes of a Nav1.7 and Nav.1.8 sodium channel blocker called PF-05089771. The diabetes trial combines PF-05089771 with Lyrica people. That trial completed this year.
- Pfizer is also studying an oral Nav 1.8 channel blocker called PF-01247324 that reduces sensory neuron excitability in rat and human neurons.
- Abbot laboratory’s A-887826 sodium channel blocker reduced allodynia in rats.
- A high throughput screening study may have uncovered a novel class of Nav1.7 inhibitors.
- In other news sodium channel gene mutations (SCN9A) were recently associated with dysautonomia and one type of small fiber neuropathy. A very large study (1500 people) will soon begin to assess the role genetic mutations in sodium channel genes play in producing chronic pain.
Conclusion

Time will tell whether sodium channel blocking drugs are the next big thing for pain.
In the summary remarks Cummins focused on the complexity of sodium channels, stated there’s alot we don’t know and that they present a lot of promise. Genentech’s representative David Hackos was the most enthusiastic. He agreed the sodium channels were very complex but felt they’d learned how to manipulate these channels pharmacologically in a very selective way (a big deal with drugs). He looked for a lot of progress in the future – and hopefully drugs.
Old drugs present the best possibilities for people in pain now. New sodium channel affecting drugs don’t appear to be a year or two years off but five or more years off. The good news is that drug companies recognize the enormous dividends that will befall any company able to deliver a good pain drug, and they appear to be investing heavily in sodium channel drugs.



With such a large publicity campaign the government has running now (either blindly or foolishly) reporting on opioid abuse, coupled with the fact that they are ignoring the statistics of responsible opioid patients, how will those who depend on these older drugs to live, get through this 5-year timeframe? Will opioids continue to be available before new drugs can be made, or will they be outlawed soon, leaving pain patients in catastrophic states of physical and mind? Will physicians and pharmacists continue to be so strapped by the fear of the DEA that they will lose their licenses, be given a little room to help their patients?
I still swear by my taking a cannabinoid called nabilone or cesamet (as well as cymbalta) for pain relief. I used to be on fentenyl (100 mg patches-a huge amount) and long acting morphine. I became tolerant, not addicted, the mu receptors for opioids were “plugged up” so no increase in these drugs helped at all, they basically just passed through me and my pain got worse and worse.
My new GP was very concerned about the amount of narcotics I had each day and referred me to an addiction specialist, which at first I wasn’t happy about-I didn’t think anything other meds would help, gabapentin and lyrica made me worse. So I slowly, slowly got off all narcotics going from fentenyl to long acting morphine to short-acting etc. In the meantime I started taking cesamet and it worked extremely wel. Two years later I was taking the max dosage and was starting to have pain again, I then added cymbalta and for the past 3 years or so this still keeps the pain manageable.
I’ve only had Fibromyalgia for a year belong to a group & educated myself to differ the standard D vitamins to include others since doing so I’m getting periods of remission am I Likely to too clear this disability others I know don’t get frequent remission. I was in the beginning put onto Gaboline for a trial it helped but after determination in losing weight I no longer wanted the medication I was switched to duxeline fet no benefit I master my own vitamins & changed my diet to the best advantage of my quality of life with Fibromyalgia as I’m still in early stages is there any hope of this disability fading
I was happily surprised to discover, at least in my experience after 7 yrs of fibro (or FUBAR) that my answer came from within, not outside of me. I believe I developed fibro after several different experiences of extreme emotional trauma, beginning at age 5. My body just shut down within 24 hours. Not the typical onset of fibro, but 3 Drs agreed on the DX of fibro.
No drug decreased the intensity of my pain, or it worked temporarily then stopped working. so within a year of onset and after months of deliberation, I asked my PCP if oxycodone might help. I’d been his patient for 15 yrs, he knew I preferred not taking any pharma drugs. He knew me as a healthy patient for years and by 2012, a patient with a debilitating disorder. He made it clear how addictive Oxy was. At the time, I felt if there was no cure, I at least wanted to lead some semblance of a “life.” Oxycodone enabled me to get out of bed. I didn’t go anywhere, but I finally got relief.
I spent YEARS incessantly researching alternative healing modalities, since western medicine offered nothing but pain meds. I tried several different modalities and was coming closer to decreasing the horrific pain. Thank God for my Husband and what a blessing he has been as my advocate!
What finally triggered my first wave of healing was a QHHT (Quantum Healing Hypnosis Technique) session, in May 2018. Hubby and I began the Keto eating lifestyle on 7/23/18 and dropped 30 lbs each and have easily maintained. I received another healing boost in November after a “Life Between Lives” session. The “LBL” session required a much longer hypnosis induction time, however my certified practitioner had to regress me back to the womb and prior to the womb before I incarnated. Desperate times call for desperate measures.
After I processed that session the pain decreased significantly. My pain was completely controlled other than an occasional flare after grocery shopping. After 3 consecutive months of completely controlled pain, I felt ready to “try” to begin the weaning process with oxycodone.
It’s only been a week, but it’s going well. I’m doing this process slowly to attempt to avoid withdrawal symptoms as much as is possible. This has worked for me. I don’t know if you are open to try anything to regain your health, or at the very least decrease the severity of this life stealing disorder.
Regardless of your belief system, I feel these sessions helped me purge old traumas I had buried that were hurting me and manifesting in chronic illness and many questions I’d had were answered. I feel I am on the path back to health.I believe our bodies have the ability to heal themselves. With those beliefs, I feel I gave my mind and body the release that was needed.
I strongly believe we should try any healing modality that intuitively feels right for us. I wish all those who suffer greatly from fibromyalgia the ability to find their path back. I absolutely believe it is possible.
Many blessings to all with fibromyalgia and every other chronic illness.
Many pathways to wellness Lynne. I’m glad you found yours and shared it with HR. Good luck on your continuing road to healing.