


“It’s the cellular equivalent of chronic fatigue.” Paul Fisher
One of the really nice things about the Emerge conference was the nice, big chunks of time they gave to some of their presenters. Paul Fisher, an Australian specialist in neurodegenerative diseases and the mitochondria, used that time to his advantage: he gave one of the most interesting chronic fatigue syndrome (ME/CFS) presentations I’ve seen in years.

Paul Fisher wanted to study ME/CFS ten years ago… (from https://www.latrobe.edu.au/our-work/parkinsons/from-accident-to-medical-breakthrough)
Fisher said he’s been knocking on ME/CFS’s door, trying to get in for over ten years. Only recently did he get his opportunity – and he’s made the best of it. My guess is that more “Paul Fishers” are out there just waiting for their opportunity to get their Seahorse machines, their brain scans, their flow cytometers and mass spectrometers, etc. to work on this fascinating and difficult disease.
Fisher, well-published and funded for work into Parkinson’s disease by the Michael J. Fox Foundation, is definitely the kind of researcher we want to attract. His interest is encouraging. Researchers of his stature don’t take on diseases without good reason, without those diseases piquing their curiosity. Why would you use your precious time on something you don’t think will work out?
A Little Crisis – A Little Rant
Which brings up my underground legitimacy crisis – the fact that I’m still somewhat shocked when someone like Fisher is interested in studying this disease. I’m like: “Really? Us? Are you sure? Did you mistake us for someone else? If I pinch myself will you go away?”

I’m acting like someone who’s been beaten on a regular basis for twenty years. No self esteem left. It’s pitiful. Decades of poor funding and neglect have left their marks.
I declare that that’s over for me, though. Instead, I take the stand that this disease is an immense opportunity that’s waiting to happen for anyone smart enough and bold enough to take it on.

I’m taking a stand – researchers should jump at the opportunity to study such a powerful and fascinating disease.
ME/CFS research is packed full of interesting findings, but that’s not where I draw this disease’s legitimacy from. I draw it from its ability to take my wonderfully athletic body and reduce it to this pitiful shadow of itself. I take it from its ability to push my incredibly smart and resourceful partner out of the workforce for the last 15 years! I take it from all our incredible, incredible stories.
This disease holds the keys to so much. It must! How could something that brings healthy, vital people in the prime of their lives down not hold within itself an immense opportunity?
We’re not a burden. We don’t need anyone’s pity. No! We are an immense opportunity. We’re offering a window into some of the fundamental processes that govern health. We offer researchers the opportunity to really break new ground – to be leaders. Don’t pass it up! These chances don’t come every day.
To those researchers who pass on us – too bad for you! The truth is we probably don’t want you, anyway. Small thinkers, people striving to fit in – people whose foremost goal is to make a career out of research – please stay away! You’ll just use up our precious resources.
Don’t forget who we are. We’re masters at slipping through the cracks! We’re paragons of paradox. We excel in breaking the mold. If that kind of thing doesn’t excite you, you should go elsewhere.
If you are excited, though, by a disease that requires you to stretch your mind, think differently, connect dots that haven’t been connected before and entertain possibilities that haven’t been entertained before – by all means, please apply. You’ll find a community that will support you like no other.
Fishing for Mitochondrial Problems in ME/CFS (And Finding Them)
Note that Fisher is studying the mitochondria outside of the plasma – where some sort of inhibiting factor may reside. If anything in the blood is interfering with mitochondrial production – as Ron Davis and Oystein Fluge have suggested – Fisher’s tests will not pick it up.
On the other hand, there’s a purity to Fisher’s examination of just the mitochondria. No outside influences allowed; that is, unless the mitochondria came into the experiment already damaged by something in the blood – a possibility.
Fisher immortalized ME/CFS lymphocytes so that he could grow them in vitro (in the lab) and test (i.e. torture) them again and again and again. Fisher put ME/CFS mitochondria through the wringer so many times that one almost ends up feeling sorry for them.
The first thing Fisher wanted to get across is that mitochondrial respiration – the act of producing energy – is VERY complex, involving numerous pathways. The act of measuring the mitochondria’s ability to produce energy, however, is actually quite simple. Since they run on oxygen, you simply need to measure the amount of oxygen they consume in order to determine how much energy they’re producing.
You might think that producing energy would be – should be – must be – a clean process. After all, you are dealing with something that could potentially go “boom”. (Look at Chernobyl.) Energy production, however, is inherently volatile, and like any volatile situation, it’s a bit hard to control.
Some of the electrons involved in the energy production process inevitably leak out of the mitochondria, create reactive oxygen species (ROS), and attempt to tear apart any cell they come into contact with. Energy production is essential, but it’s also the biggest producer of free radicals in our bodies.
Fisher was merciless in his efforts to torture, er test, the mitochondria of the immortalized ME/CFS white blood cells in every which way he could. He inhibited every complex (there are five of them) of the Krebs cycle possible; plus, he assessed how well glycolysis – the process of turning glucose into ATP for the mitochondria – was doing.
Results
Normal
As expected, the mitochondrial mass and the number of mitochondrial genome copies was normal: there was no drop in number or mass of mitochondria in ME/CFS. The surprise came in the glycolysis findings – all normal! Glycolytic rate, glycolytic production, capacity, reserve – all perfectly fine.
That, of course, bucks with Neil McGregor’s recent findings and hypothesis that ME/CFS begins and ends with glycolytic issues. I asked McGregor what was up with this? He believes that Fisher’s inability to use plasma (the Seahorse will not accept it) could account for the different results.
Fisher also found that the rate of ATP synthesis and production and ROS production were within normal limits. The cells appeared to be producing normal amounts of ATP and were not being slammed, as has been suspected, by reactive oxygen species or free radicals.
Major Mitochondrial Complex Hit Hard
However, then the results began playing a familiar tune.
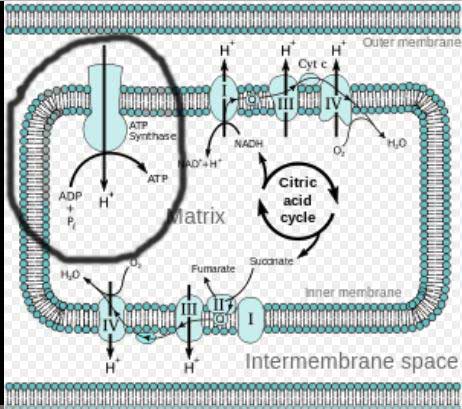
Complex V highlighted – Electron transport chain in mitochondria. Note that this appears to be the only complex where ATP is actually created.
ATP production was normal, but once Fisher began stressing the energy production systems strange things began happening. In particular, Fisher found that Complex V – the last of the five complexes and arguably the most important – responded poorly. Complex V transfers protons through the inner mitochondrial membranes to the mitochondria. That process, which involves ATP synthase, releases the energy used to drive ATP synthesis.
The complex looked fine at baseline, but when put under stress it pooped out – suffering a 25% drop in production compared to healthy mitochondria.
That drop caused the other four complexes to spring into action. ME/CFS patients’ mitochondrial membranes became packed with extra copies of the mitochondrial complexes. Complex I – the main electron gradient producing complex that provides electrons to Complex V – went gangbusters in an attempt to provide Complex V with as many electrons as possible. ME/CFS patients’ proton pumps worked overtime to pump protons out of the mitochondria en masse in order to increase the membrane potential and get more electrons into the cell. Enzymes that consumed O2 were jacked up as well.
The Gist
Using the Seahorse machine, Paul Fisher, a noted mitochondrial expert, tortured the mitochondria in ME/CFS patient’s immune cells in every way possible.
Note that Fisher (and every researcher who uses the Seahorse machine) must take the cells out of the plasma – where an inhibitory factor may be present – to test them.
Energy production at rest was normal but when put under stress the cells failed to respond.
Fisher’s key finding was a reduction in Complex V activity in the mitochondria. Complex is the last complex in the electron transport cycle – it’s one from which ATP is released.
Fisher also found evidence that the other mitochondrial complexes and other processes in the cell were being upregulated in an attempt to compensate for Complex V’s deterioration.
Glycolysis appeared to be normal but Fisher found evidence of a switch from burning glucose for fuel to burning fatty acids.
Fisher believes the answer to the mitochondrial problems will be complex and has begun a hunt to search for the core mitochondrial issue present.
More studies are needed but several Seahorse studies have found that the mitochondria in the immune cells of people with ME/CFS are been unable to adequately respond to stress.
The Complex I upregulation detected by the Seahorse was validated when a proteomic analysis demonstrated that a huge increase in Complex I proteins had taken place.
The end result was that the ME/CFS mitochondria were working harder than ever. They were able to get the cells’ ATP production up to normal – at rest – but not enough to handle stressful conditions. The fact that mitochondrial measures generally correlated significantly with disease severity in ME/CFS (i.e. were worse in the patients with the most severe disease) was encouraging.
While Fisher did not find problems with glycolysis, he did find that the energy stressed ME/CFS patients’ cells had moved from breaking down glucose to breaking down the more ATP-rich fatty acids in order to squeeze out as much energy as possible. That fatty acid switch – which was similar to what McGregor found (but which McGregor extended to proteins) – was demonstrated by an increase in levels of the six enzymes that break down the fatty acids, as well as the proteins that import fatty acids into the cells.
The good news is that the problem is not subtle: Fisher appears to be seeing major problems across the mitochondrial pathways.
The bad news is how complex the mitochondria are. Fisher doesn’t think he’s going to find a single cause that responds to a single, simple fix. He also left open the possibility that the Complex V problems could be a consequence of another as yet unknown problem.
Mitochondrial Results Starting to Add Up
We haven’t had a lot of mitochondrial studies, but the results are beginning to add up. Tomas’s 2018 results could have been taken word for word from Fisher’s presentation. Using a Seahorse machine, the U.K. group found that the mitochondria pooped out when put under stress as well. Tate recently reported getting similar results in his New Zealand cohort as well.
“Maximal respiration was determined to be the key parameter in mitochondrial function to differ between CFS and control PBMCs….The lower maximal respiration in CFS PBMCs suggests that when the cells experience physiological stress they are less able to elevate their respiration rate to compensate for the increase in stress and are unable to fulfill cellular energy demands.”
We need more and larger studies, but three studies thousands of miles apart getting similar results using the same machine is pretty darn good. It should be noted that Myhill also found mitochondrial dysfunction in every patient tested.
A 2016 Stanford study, which did not use the Seahorse machine, might seem to be an outlier, but maybe not. It found increased ATP respiration – but from non-mitochondrial sources – and increased mitochondrial membrane area and increased number of cristae in ME/CFS, two findings which might be compensatory mechanisms as well.
Plus, in 2015 an in-silico model of ME/CFS showed how problems with mitochondrial functioning could potentially explain the post-exertional problems and long recovery periods needed. The authors suggested a number of issues, including mitochondrial deletions, Epstein-Barr virus-induced alterations of mitochondrial gene transcription, pro-inflammatory cytokines and increased levels of oxidative stress, that could result in an inability to ramp energy levels up in ME/CFS.
With Solve ME’s Ramsay award and other studies underway that are measuring energy production in immune cells, expect more results soon.
Meanwhile, Fisher is engaged and on the hunt. His next step – which he stated he is taking now – is to manipulate one thing at a time in an attempt to identify the core problem in the mitochondria. His first ME/CFS paper should be published soon.



Thanks Cort. Your “little rant” was very encouraging!
“The complex looked fine at baseline, but when put under stress it pooped out…”
I wonder if the baseline vs. stress difference might account for why different patients, or even the same patient during different symptom phases, can experience contradictory reactions to supplements designed to increase ATP production.
For example, when I tried taking NT Factor Energy, I found that after a few days it always seemed to increase the wired and tired symptoms, leading to sleep loss and eventual crash. I’ve had similar reactions to various doses of CoQ10/Ubiquinol and MCT oil. But I wonder if I would respond more positively had I tried them during extended crash phases, when I’m cold, sleeping long hours, etc.
(A lot of this article was over my head, so please forgive me if this is oversimplified.)
🙂
I’ve actually had similar experiences of energy producing treatments that started off making me feel better but ultimately left me crashed….I’ve wondered if the energy production process is proceeding but resulting in some toxic byproduct. Naviaux is interesting in this respect as I’ve seen him demonstrate that if X pathway is blocked – this will buildup.
With regard to this article I can only imagine that force-feeding one aspect of energy production in the mitochondria might lead to problem elsewhere. What happens to the excess? Does it show up in increased oxidative stress?
Somehow this disease is kept intact….
I think trying energy supplements when you’re in a crash is a good idea.
I’ve had CFS for many, many years. About 20 years ago I was trying different things. One was traditional Chinese herbal medicine, and the other was a psychic healer.
Both of them worked, but only briefly — just like Cort’s experience.
The first round or two of (boiling etc. then drinking tea) of the herbal remedy worked great . . . and then I crashed.
Couple years later, tried the psychic healer. She was fine. Earnest, serious — and it really worked. The first time or two.
For both of them, the effect was like revving up an engine where something is gumming up the works. As long as it’s just idling, the “gummed up” quality is not noticeable. But try to get real performance, and it all gets jammed up.
– – – – –
And p.s. to Cort:
You said, “I think trying energy supplements when you’re in a crash is a good idea.”
Did you actually mean not a good idea?
The only problem with anaerobic capacity in PwMEs is that it’s the only part of our metabolism that’s working correctly. Most retain short term strength precisely because we can use the 2 minutes worth of glycogen and creatine phosphate stored in the muscles. It’s the lack of O2 and replacement glycogen after the 2 minute mark that causes problems. Recall Klimas describing how patients went into “anaerobic threshold” after only 2-3 minutes? That’s why. Once the O2 and replacement glycogen are not forthcoming, it forces the muscles back to the muscle stored glycogen which was mostly used up in the first 2 minutes. The result is over production and accumulation of metabolic byproducts which results in rapidly increasing muscle stiffness and fatiguing. Studies have shown that the muscles in PwMEs are metabolizing ADP and AMP and the resulting metabolic byproducts are non-recyclable. I figured out long ago that if there’s a lack of blood flow entering the muscle cell, there’s a correlary lack of blood exiting the cell; which is what carries the metabolic byproducts out of the cell. Hence the over production and over accumulation of the lactic and phosphoric acid in the muscles.
@Keir It’s true that moderate patients have ability to engage in very high intensity for a few minutes at a time. The problem is that such exertion will surely bring about PEM the next day. (Sustained high intensity exertion has been THE surest way to trigger PEM in my experience). Severely ill patients of course, being constantly in PEM state, wouldn’t be able to.
Note that most CFS patients are severely deconditioned. Quickly hitting the anaerobic threshold may have to do with that rather than any abnormality of CFS itself. Keeping the deconditioning aspect of CFS in mind, and PEM being a sickness, may keep researchers from going down the wrong path too far.
Thanks for the “rant” Cort. I am currently suffering from yet another round of even doubting my own reality in response to attempting to justify being unable to keep a commitment to do something that would wonderful and fun (if I were not ill). After all these years and so many serious crashes I still feel the pressure to do something that I shouldn’t because someone else really thinks that I “can” if I tried. If that makes sense. I certainly appreciate all of your efforts on behalf of all of us.
Thanks Cort. I second this comment, I enjoyed your article and how much fun you were having with it. ??Brightened my day….
I echo these comments. We need to stand up (in our spirit if not in our body!) and KNOW we have a legitimate illness, and that we are right to look after ourselves in ways that keep us as well as possible. Often we are the only judges of that. It seems all of us need to find calm but firm ways of dealing with well-meaning ‘encouragement’!
Learning how resist ‘being made to feel a fraud/guilty’ for declining opportunities is SO important. Accepting poor treatment/abuse/neglect/disbelief has to end. It’s hard to be strong in spirit when you’re weak in body (and often in brain power too) but we need to know we are standing together. Not to be aggressive, but to be assertive.
I love Cort’s statement,
“I declare that that’s over for me, though. Instead, I take the stand that this disease is an immense opportunity that’s waiting to happen for anyone smart enough and bold enough to take it on.”
Thank you Cort and thank you to the Health Rising community.
Many people including myself have literally cured ourselves or at least can overcome the fatigue quickly that might otherwise keep us in the house all day. The Gupta Program ;concentrates on retraining the amygdala responsible for the fight or flight response. There is a DVD program that is worth 20x its cost. I urge anyone suffering from ME/CFS to check it out.! A life saver literally!
Cort Johnson, I wonder if his findings similar to Complex V Deficiency can go with type V Glycogen Storage Disease it is the one besides type XIII that both have exercise
Intolerance & type V known as McArdle Disease is an illness that is found in countless told they have Chronic Fatigue Syndrome. I wonder if this Researcher to
fast forward his work could also look at the blood of McArdle type V patients which would be easy to do…He should also run panels on these bloods for GSD types it is
an 18 panel done or he could look for GSD V & GSD XIII by themselves it is cheaper & he also could do muscle biopsies looking for the GSD V & XIII abnormalities they
are easy to test for now aside from Genetics…The full GSD panel on the NIH in the UK cost them to run the 18 panel is £900.00 there is also a lab in Europe that runs it
cheaper not sure where though…I had an ultrasound of Heart they say I have (LVH) Left Ventricular Hypertrophy, I did searches the other day it can also be involved in
Eagle Syndrome which my Dental Panoramic is showing I likely have now & will likely need Surgery both sides I am soon to get a CT Contrast Scan to confirm the
length involved of these bones calcifications…
Thank you Cort! These articles give me so much hope! Now if I could just stay away from “stressing” my mitochondria. :-).
I related strongly to this article, as many years ago (1995?) a Dr in Germany explained the cause of my many symptoms to be “ mitochondriapathy”. This diagnosis was met with blank stares from my own Drs here at home, and I am still struggling with muscle weakness, fatigue, brain fog, sleep problems, etc. It will be interesting to see if there are future studies in mitochondrial disfunction re ME/CFS. Prior to being diagnosed with fibromyalgia, I was diagnosed with E-B virus, and CFS.
So based on these findings, and if the disease is considered mitochondria dysfunction, do we no longer call it neurological inflammation? Or is it both?
I would guess both.
We still need more studies but if the mitochondria are messed up – they could be causing all sorts of problem. Fisher was studying immune cells – mitochondrial problems in the immune cells could cause all sorts of problems, including, I would guess – is inflammation since one possible outcome of mitochondrial disruption is increased oxidative stress.
Plus a pathogen could be tweaking the mitochondria or they could be in the dauer state Naviaux has hypothesized or a toxin or poor blood flows…These mitochondria were isolated but Ron Davis has speculated that mitochondria from ME/CFS patient’s could have already been damaged by something in the blood.
If the mitochondria are not working well the cells they are found in will not be working well.
Hi I am wondering if anyone has tried anti-viral treatments for the Epstein Barr Virus (EBV) or herpes virus 6, which seems to be the cause of the CFS and mitochondria dysfunction.
Many people have and for some people they work! (For others they don’t). They can take up to a year or even more to work in some people.
I’ve read about these (on Healthrising!) before but they still seem odd to me, both claims:
“The cells appeared to be producing normal amounts of ATP and were not being slammed, as has been suspected, by reactive oxygen species or free radicals.”
The lack of oxidative stress doesn’t fit my believes nor how I interpret my experiences.
To picture a recent experience: when over-exhausting for a few weeks a piece (both as an “experiment” for being able to better observe how symptoms change over time and a plain temporal lapse in discipline to stay rigorous within my energy envelope) I started to have a nasty skin rash on a few limited places where I had vitiligo since long before I got ME. The rash kinda moved outer wards from it starting center and left a clean-ish circle with slightly different color compared to the original surrounded with an small ring of small individual round dots of rash with skin that somewhat peeled of.
When continuing the “experiment” I developed another dot that was about 1 cm in diameter and that was red with a slight touch of blue. After taking a bath that skin seemed like the very thin upper layer of it was peeled of but the 1 cm diameter region still was sticking a bit up above the other skin. It was less then a single mm high but it appeared inflamed. It looked in between how an abrasion (wound one gets when a rough object peels a tiny patch of skin if I got the translation correct) and a fresh wound from a strong acid looks like.
Now vitiligo has a strong relationship with too strong immunity. Pushing my exertion envelope for too long caused some patches of skin too be eaten away by my immune system! And my skin didn’t really participate in the exhaustion. Imagine what it could do with tissue like muscle, fascia or brain cells being at the center of that exhaustion! Or my gut tissue flaring up in pain. For now I learned enough to go back to the envelope as I disliked the mental picture of these regions with tissue eaten away in my gut, muscle, brain… :-).
To get back to the topic: A strong immune reaction goes hand in hand with high amounts of reactive oxygen species and vitiligo is often said to go hand in hand with high oxidative stress. See for example http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3068765/.
So I’ll still try to get back to my “oxidative stress is still a major thing IMO” mantra.
I wouldn’t be surprised at all if oxidative stress ends up playing a major role in ME/CFS.
Your rash sounds like my (diagnosed by a Dermatologist/skin cancer specialist) Pigmented Purpuris Dermatosis on the front of my shins. As the reddish flare-up dies down, it is replaced by a brownish ‘scar’ (which is permanent). Some of the larger patches appear to have a top layer that peels off.
I have what appears to be a different rash on my left inner ankle that at one stage was all under my foot arch too. It fades more and leaves only a faint skin discolouration. A couple of times I had thought this ankle rash flared more when I eat dairy products (I am lactose intolerant) or was severely stressed. I see an asthma/allergy/immunology specialist on the 6th August as I have all the signs of MCAS (Mast Cell Activation Syndrome).
Yes, that sounds so much like it. There is indeed some brownish scaring (a bit “cloud like” with round borders) but also some bleaching at other places. Maybe that’s the result of one process or it are two different processes happening in the same region?
If I interpret it correctly I saw several pictures of vitiligo with white areas surrounded by something resembling a brownish border. See the second picture, fingers in en.wikipedia.org/wiki/Vitiligo. I don’t think those parts are just normal darker skin.
Last time I read “The complex looked fine at baseline, but when put under stress it pooped out – suffering a 25% drop in production compared to healthy mitochondria.”
It didn’t fully convince me. We can do WAY less then 75% of healthy people. The body *should* be able to still do plenty even with only 75% of the maximum present. When a healthy person can run for an hour at twice the speed of walking with 100% energy production, I should be able to walk at normal speed for an hour with 75% of that production. Or at the very least 10 minutes at two thirds of normal walking speed. 75% of normal seems not that much more then a deviation between strong / average / weaker people who are still not ill.
I also have difficulties with jumping from this 75% of normal (at exercise!) to
“That drop caused the other four complexes to spring into action. ME/CFS patients’ mitochondrial membranes became packed with extra copies of the mitochondrial complexes. Complex 1 – the main electron gradient producing complex that provides electrons to Complex V – went gangbusters in an attempt to provide Complex V with as many electrons as possible. ME/CFS patients’ proton pumps worked overtime to pump protons out of the mitochondria en masse in order to increase the membrane potential and get more electrons into the cell.”
In an ideal mathematical situation if one can only produce 75% in one part of the chain, one should up another part of the chain to “compensate back the 75% to 100%”. With that I do mean: if the other complexes up there output with one third they should be able to compensate for it. That is at least assuming the reaction speed of complex V is proportional to both the amount of complex V and the amount of protons present. I know, it’s an oversimplification but still I see few reason to go “gangbusters” on the compensation mechanism. Remember, even if far more compensation was needed, why on Earth do we need the last 25% percent of ATP production to get from our bed to the bathroom??? A compensation to let’s say 80, 85, 90% of normal ATP production should suffice and that would not need the other complexes to go gangbusters wouldn’t you thing???
OK! How might this figure in?
Most of the energy the body produces goes to keep it running. The next top energy user is digestion. Running around, playing tennis, climbing the Matterhorn, etc. – If I remember correctly all that uses up something like 10-15% of our energy.
So if you whack energy production by 25% maybe you really are cutting into activity (???)
Hi Cort,
That 10 to 15% of energy for activities seems to be way off my estimates. At best it is an average and not one including climbing the Matterhorn.
Here is my back of the envelope calculation.
Assume a 2000 kCal a day diet. 1cal equals 4.2 Joule. 2000 kCal equals 8400 kJ. 1 Wh, or 1 Watt sustained for 1 hour, equals 3600 Joule or 3.6 KJ.
That makes 8400 kJ roughly 2400 Wh. Divided by 24 hours a day that gives us 100 Watt average fuel available to do things IF we would digest 100% of our food. That is an optimistic estimate.
Now a young healthy adult can bicycle quite some time producing 100 Watt of *mechanical* power. As the conversion from glucose and fat to mechanical power is far less then 100% efficient, that requires a lot more power. I think a figure between 25 and 40% efficiency is realistic as far as I recall.
Let us assume 40%, a quite high value that gives us the lowest estimates for how much extra “fuel” producing 100 Watt on a bicycle consumes. That would be 100 Watt * 100% / 40% = 250 Watt of fuel (glucose, fat…). And that is just on top of basic needs for breathing, hard beat, liver function, consciousness…
Let us assume this basic needs are a bit below the 100 Watt average available day round, take 75 Watt for basic metabolism.
This not that extreme bicycling would up “fuel” consumption from 75 Watt to 75 + 250 = 325 Watt or bicycling “this much” would cause an increase of over 300% in fuel consumption over basic metabolism. And a trained bicyclist let alone a professional one would go way beyond 100 Watts of mechanical produced power for over an hour in time.
To put it in context, at the beginning of my disease but still better times I went to a revalidation center doing physical exercise. One of the homes trainers was “self powered” meaning it derived the electricity it needed from the person bicycling on it. The catch was that it required IIRC 30 Watt minimal to function or it would stutter and block.
The week after a flu vaccination the machine blocked like five times in five minutes before I gave up because I couldn’t deliver this minimum required power to the machine. A friendly 75 year old chap revalidating form IIRC hart surgery happily could keep the machine spinning…. …and that was still at better times.
At my worst period it would have been every day very exhausting and challenging to reach the home trainer by crossing the 20 meter (about 22 yard) distance to it. At average days I would be extremely challenging and at worst day I may or may not have reached it crawling on hand and feet. If my mitochondria performed “only” at 75% of max I doubt that would ever have happened. Just as much as I doubt lifting a fork with a minimal amount of food on it to get it to my mouth should have been so often an exhausting task. That is sooooo close to basic metabolism in my opinion…
Here is the part I try and cram “mitochondria normal at rest (out of plasma)”, “normal amounts of ATP and oxidative stress (out of plasma)”, “drop of 25% to 75% of complex V production at exertion”, “enormous increase in other complexes and even worse in complex I”, “devastating exhaustion” and “ridiculous high oxidative stress” together.
Let me start with
“Complex 1 – the main electron gradient producing complex that provides electrons to Complex V – went gangbusters in an attempt to provide Complex V with as many electrons as possible. ME/CFS patients’ proton pumps worked overtime to pump protons out of the mitochondria en masse in order to increase the membrane potential and get more electrons into the cell.”
It seems these complexes including complex 1 produce far more then a 33% increase in protons to pump out of the membrane “in order to increase membrane potential and get more electrons into the cell”. But even if it were “just” an increase of 33%, then both the membrane potential (amount of voltage) and concentration of protons (H+) should be “abnormally high”. That in itself should cause plenty of problems itself as this seems quite an imbalanced state.
OR at least I thought so at first. Creating such high amounts of H+ surrounding the mitochondria would almost create the opposite of oxidative stress just outside the border of the mitochondria. As much as an abundance of charged oxygen or OH(-) creates plenty of oxidative stress, it *feels* like an abundance of H+ surrounding the mitochondria creates an abundance of what I would call “reductive” stress (with reduction the opposite of oxidation).
Now that supposed massive “reductive stress” might form a formidable shield canceling the potential oxidative stress attacking the mitochondria. I have no solid proof yet, but my limited knowledge of basic chemistry says that OH- , OH, O2- should react (very?) fast (“violently”) with the abundance of H+ forming a shield around the mitochondria. That, in effect would be a !deliberate and vital! defense of the mitochondria against a massive outside attack by oxidative stress.
If so, not having increased oxidative stress *in* the mitochondria (out of plasma) would point to massive oxidative stress coming from either other parts of the cell or from the environment / plasma. Not having increased oxidative stress would in fact point to a super-oxidative storm. One that would be hard to catch as the mitochondria are NOT turned into factories of oxidative stress (hydrogen peroxide) but into massive factories of “oxidative stress compensation chemicals (H+)”. That would add the concept of “reductive stress shielding of mitochondria” on top of the “oxidative stress shielding” concept of Naviaux.
It would also help explain why a “modest” decrease in complex V productivity (“only” 25% and only when at exertion) drives the other complexes to produces SOOOOO much more protons: they get eaten away by the oxidative stress attacking them, thereby performing their role as a shield against oxidative stress.
That this hypothetical process still happens when the cells are out of the plasma could point to different options:
A) Other parts of the cell produce still plenty of oxidative stress in these cells, which the mitochondria defend against. That could be for example the protein producing parts producing far too much misfolded proteins that need to be destructed by… …oxidative stress if refolding them a few times fails.
B) Another option: the chemicals enticing the mitochondria to produce an abundance of protons still linger in those cells and the cells in fact “dissipate” excess protons by “reductive stress” modifying chemicals in the cell by absorbing protons, a bit like oxidative stress is dissipated by modifying chemicals and binding to them.
That the crippling of complex V does not happen at rest does point to option A) being at least part of the problem. If the basic chemicals producing excess H+ were still an abundant remain in the cells then the effect would still take place at rest. Of coarse there is still the option that “exertion sensing chemicals” are still abundant that produce the chemicals that steer towards too high H+ production at exercise, but that is a (possible and more difficult) second order effect. Now these “other parts” producing too much oxidative stress however still seem to be increased a lot by putting these cells in ME patients plasma.
Could that mean that a perceived outside danger is enticing other parts of the cell then mitochondria to produce excessive oxidative stress and that the mitochondria in turn defend themselves against that? Maybe a hypothesis on top of a hypothesis is getting us too far…
As far as I know of, it’s a radical new concept but it could explain a few things.
For example, if ones body were more keen to produce this hypothetical “reductive stress” or in other words to “like to” produce higher quantities of protons and putting them on the outer mitochondria membrane wall, then having a high oxidative stress outside event (like the immune system producing plenty of ROS fighting of pathogens) would trigger the mitochondria to go “gangbusters” on producing protons to maintain there desired high amount of protons at the outer shell.
So the mitochondria could both have to resort to producing far more protons in order to compensate for a deficient complex V, or they could cripple complex V in order to keep sufficient protons for their functioning or more like for their shielding against oxidative stress (as crippling complex V means fewer protons are “consumed” by the mitochondria to form ATP).
The stuff below lead to the ideas in the previous comment; posted them here for those trying to follow (and correct, improve or denounce) my ideas and dig deeper but I left them out of the main comment as they are complex and confusing, at least to me.
When looking up “mitochondrial ATP synthase ROS” then I did found sciencedirect.com/science/article/pii/S0005272814000978 saying:
“Mitochondria are main sources of ROS production.”
“ROS are signaling molecules that can regulate cell death or cell survival.”
“Inhibition of the H+-ATP synthase regulates mitochondrial ROS production.”
=> note that “regulates” fails to say which direction, paper after paywall but stomping it could be in order to do so.
IF1 inhibits the synthase activity of the H+-ATP synthase in human carcinomas.
“These findings are further placed into the physiological context to stress the emerging roles played by IF1 in metabolic reprogramming in cancer, in hypoxia and in cellular differentiation.”
Hypoxia? Seems to be a companion of ME…
“Special emphasis is given to the role of IF1 as driver of the generation of a reactive oxygen species signal that, emanating from mitochondria, is able to reprogram the nucleus of the cell to confer by various signaling pathways a cell-death resistant phenotype against oxidative stress.”
Now one thing that goes with Naviaux’s Dauer concept is inhibition. That involves low cell replacement rates (normal cell dying rates with low replacement rates is a very poor form of inhibition lasting very shortly only IMO). And that involves low cell death rates. IF1 and with it ATP synthase seems to be directly involved in delaying cell death that is IMO essential for prolonged inhibition.
ncbi.nlm.nih.gov/pmc/articles/PMC2919093/ says something incomprehensible as
“Overexpression of IF1 or of its pH-insensitive H49K mutant in cells that express low levels of IF1 triggers the up-regulation of aerobic glycolysis and the inhibition of oxidative phosphorylation with concurrent mitochondrial hyperpolarization.”
Now the latter seems to say “produce plenty of protons that don’t all get destroyed by oxidative stress”. But how more aerobic glycolysis goes hand in hand with inhibition of oxidative phosphorylation is unclear to me.
“The ATPase Inhibitory Factor 1 promotes metabolic rewiring to an enhanced aerobic glycolysis and the subsequent production of mitochondrial reactive oxygen species. The generated reactive oxygen species are able to reprogram the nucleus to support tumor development by arresting cell death. Overall, we discuss the cross-talk between reactive oxygen species signaling and mitochondrial function that is crucial in determining the cellular fate”
Made no sense to me. The generated reactive oxygen species reprogram the nucleus by arresting cell death (that IMO is going into the final stage by having the cell killed by massive ROS release and the immune cell attacking it)? Can’t wrap my mind around it. I probably don’t understand all but it sounds like some amount of increased oxidative stress prevents the cell dying from massive oxidative stress. If so, that seems a very unstable situation to me, something I don’t see nature picking as a main route to life. That triggered the idea that oxidative stress is not triggering the mitochondria to produce ROS but to produce a “proton shield” instead.
Searching further into “H+-ATP synthase” led me too
ncbi.nlm.nih.gov/pmc/articles/PMC3936247/ with title “Redox-regulation of mitochondrial ATP synthase”
The title itself says that mitochondrial ATP synthase is directly regulated by the amount of oxidative stress (or the lack thereof or even the hypothetical reductive stress).
“Reversible cysteine oxidative post-translational modifications (Ox-PTMs) represent an important mechanism to regulate protein structure and function. In mitochondria, redox-reactions can modulate components of the electron transport chain (ETC), the F1FO-ATP synthase complex and other matrix proteins/enzymes.”
“Among this group, there is a selected subset of amino acid residues that can function as redox-switches. These unique sites are proposed to monitor the cell’s oxidative balance through their response to the various Ox-PTMs. In this review, the role of Ox-PTMs in the regulation of the F1FO-ATP synthase complex is discussed”
That basically says: at least the activity of this ATP-synthase complex (some sort of complex V) is *directly* affected by the amount of oxidative stress.
Also: this “reversible cysteine oxidative post-translational modifications” are an extension of the glutathione subsystem with IMO body wide signalling. It affects hundreds of chemicals that are flipping on and of switches according to the amount of oxidative stress and these chemicals very often interact with basic energy producing processes. My guess is that these (class of) chemicals may be a good candidate for what “is in ME blood” making ME blood trigger ME symptoms in healthy cells.
“The ETC is also a primary source of ROS/RNS production in the mitochondria which has been found to impact the F1FO-ATP synthase complex. There is considerable knowledge about the structure and function of F1FO-ATP synthase and its subunits”
There could be another option then part of the oxidative stress having to come from inside the other parts inside the cells then the mitochondria.
The body seems to love to use the difference between signals to control processes, like for example both sleep hormones and activity hormones can be either low or high but what happens depends on the difference between both. For example one can have low activity hormones but even lower sleep hormones so one stays awake or one can have high sleep hormones but even higher activity hormones and one stays awake as well.
In a same way, one chemical may “remember” past, accumulated oxidative stress and another chemical may indicate how much energy is demanded. If ATP synthase activity is depended on the difference in the respective strength of these two signals AND ATP synthase has a different sensitivity to both then mathematically that would make inhibiting or activating ATP synthase the result of where both lines with different slope cross.
All this is too say:
* both options A) and B) are still valid.
* the chemicals making ME blood special seem IMO to “remember past amounts of accumulated oxidative stress.
Hi Dejeurgen. Really interesting ideas as always. High oxidative stress is known to disrupt mitochondrial functioning and you have presented an elegant redox theory to explain how this might occur in reality. However, one incongruity sticks out to me. The cells Fisher was testing are in vitro, not in vivo. I don’t know how Fisher inhibited the mitochondria, but it does not appear that the in vitro mitochondria are mounting a reduction response to an onslaught of oxidative stress. Instead, Fisher’s findings suggest that the electron V complex has been previously damaged by some unknown agent and no longer works efficiently either in vivo or in vitro. I am inclined to think that the issues with the electron V complex that Fisher has observed represents damage because none of the science to date indicates that ME/CFS patients have a genetic mitochondrial disorder. We generally acquire the illness following some “hit and run” initiating trigger. Two researchers that I am aware of have found that the mitochondria display condensed cristae. Perhaps the mitochondria have adapted to protect themselves from some outside attack?
What I find it impossible to ascertain from the science right now is if there is some perpetuating damaging factor floating around in our blood or cells or if we have simply got stuck in a metabolic trap as Ron Davis and Robert Phair have suggested is a possibility. Time will tell.
Whenever new science is presented, most of the time all I see is vicious feedback loops. Say, a virus damages our mitochondria and we can no longer produce aerobic energy efficiently in the electron chain and need to rely more upon dirty back up forms of energy production. I have always felt like a car that is missing gears. If I go at the right speed then I am fortunate enough to be entirely comfortable. However, adrenaline is and always has been my greatest enemy. It is like putting my foot on the accelerator pedal when I am stuck in the lower gears. The engine is revving but I don’t hear it (although my blood pressure typically shoots high when I overdo it before falling normal / low again). I suppose this is a survival over-ride. If we need to escape life threatening danger then adrenaline mobilises all available energy sources. The problem though is that adrenaline regularly takes me over my modest limits and creates some dirty by-products that my body needs to clean up. Over time, I suspect that my clean-up systems have been wearing out (glutathione down) and are less able to deal with the mess (including oxidative and nitrosative) which continues to attack my brain, gut, heart, arteries, blood cells, immune system, thereby perpetuating the damage, further wearing out the clean-up systems and compounding the need for my body to shut off and bunker down. If I could just sort out my pacing then I am certain I would slowly improve but pacing requires massive discipline and sacrifice, plus life doesn’t really come with enough smooth water and society doesn’t carve us out any space to heal.
35 year sufferer here
“Simething in the blood”
Interesting to me as about a month or so ago I happened to glance at my foot and see a red line from my foot upward on my ankle!
Fast forward to last week…. I find a wildlife biologist willing to help me….he asks for pictures of my eyes
(Iridology) and sends me a text saying I have serious blood
Contamination!! I do have low red platelets
I am now sold on iridology.says he’s delt with a few cases of this and that most are resolvable. He does this out of his good heart so i know hes not a quack.
Hi debsw,
thanks for the interesting input; I’ll have to reread again (my mind is cloudy now).
“it does not appear that the in vitro mitochondria are mounting a reduction response to an onslaught of oxidative stress.”
See my response to Learner1, fits well with one of his questions.
“Instead, Fisher’s findings suggest that the electron V complex has been previously damaged by some unknown agent and no longer works efficiently either in vivo or in vitro.”
I think Fisher’s finding leave both yours, mine and other options open.
One of the nice things about my option (long lasting oxidative stress “serine altered” a key messenger switching it’s action from activating to inhibiting (or reverse) resulting finally in inhibiting of complex V), also fits well with how one can get this disease in a progressively fashion or how both marathon runners and power trainers can become prone to FM/ME (often observed).
Power trainers create very strong bouts of ATP demand lasting longer then 2 or 3 minutes the anaerobe metabolism can deliver. That creates a very rapid drain of ATP in working out cells, draining the “H+ proton shield” (in order to restore ATP as long as the workout endures) very quickly if complex V isn’t inhibited in minutes. That leaves oxidative stress free to go encountered for nearly as long as the intense workout lasts. Repeat several times a week and cell walls are thinned from the inside out.
It also helps explain the former marathon patients. As part of a long lasting sizable demand of ATP, the strength of the defensive H+ proton shield is somewhat diminished. The unbalance is far less then in the power training case. But it last for a longer time. Reduced defenses allow for a higher amount of oxidative radicals to pass through the “proton shield” slowly increase damage over time. At the end of a marathon, when oxygen/energy/blood supply is getting more troublesome or runners increase their speed seeing the chance to set a good time, the imbalance starts to increase (a lot). The end result is significant damage piled up over the course of the marathon. As cell walls start to leak, one “hits the wall”. Or maybe when metabolism is shut down just before that happens.
These ideas, just by themselves, help provide an alternative for the more and more contested lactate accumulation theory of exercise fatigue and pain. It also connects “normal fatigue” into a continuum with “devastating exhaustion” as it describes a same mechanism but starting from a vastly different starting point, making a huge difference in how much exercise becomes destructive and how well the restoration process proceeds. Same ideas hold for normal mental fatigue versus debilitating exhaustion when trying to do basic stuff in ME.
Hi Dejurgen. Thanks for your reply. Love your ideas but hope you are taking it easy and resting your brain! And yes, fatigue is clearly so much more than lactate build-up.
Given how common fatigue is, isn’t it surprising how little is truly known about it, particularly the role of the brain. If I was an ME/CFS researcher, which I most certainly am not (only the armchair kind 🙂 ), I would consider using a control group of traumatic brain injury (TBI) patients. I have known 2 unfortunate people with TBI and observed a significant overlap between their symptoms and my own. Their condition clearly results from a brain insult but also cascades through to secondary mitochondria dysfunction, in the brain at least. I still find it mind boggling that Jen Brea’s definite ME/CFS symptoms were resolved by spinal surgeries and given that the brain has not been ruled out as the source of the illness, I think it would be helpful to include TBI controls to check convergence and divergence.
I find in vitro findings like Fisher’s and Myhill et al’s helpful by separating the mitochondria from the brain, plasma and presumably any outside influences (although not the cellular fluid itself) and the mitochondria still display dysfunction. Regardless of the cause, the mitochondria are have been clearly subject to collateral damage from a disease process. I do not expect Ron Davis’s saline test to be “specific” to ME/CFS. The test will almost certainly pick up patients with genetic mitochondrial illness but it will be interesting to see if mitochondrial dysfunction shows up in other patients with conditions such as post-sepsis fatigue and cancer related fatigue. If mitochondrial dysfunction is more widespread than our own illness, this can only help to align us with the “serious” illnesses and serious research funds.
Unlike patients with TBI or post viral fatigue (often treated interchangeably as CFS but patients typically recover within 2 years), ME/CFS patients do not often achieve true recovery. Even if the brain or mitochondria are damaged in an initial “hit and run”, then surely over time the situation would improve? Brains are somewhat plastic and mitochondria are in a constant state of fusion, fission and autophagy. Discrete damage is typically weeded out. Hence, there simply must be some epigenetic change, viral presence, immune shift, dauer-type response, negative feedback loop or metabolic change that sustains the illness and continues to impact the mitochondria, as per Fisher’s findings.
Dejurgen, I 100% agree with you that oxidative stress seems like a major player in this illness. Kyle McNease’s incredible Lazarus type story chucks yet another variable into the mix – heavy metal poisoning. I find myself returning to Rich Van Konyenburg’s methylation block theory and wonder just how much credence has been given to the notion of oxidative overwhelm. We speak about oxidative stress like it is something tangible but it isn’t really – it has many, many sources (respiration and other metabolic processes, rancid oils, poor diet, heavy metals, pesticides, emotional stress, etc) and appears to be difficult to test for and quantify. Oxidative stress and low glutathione is linked with viral susceptibility and poor health outcomes, and it has been hypothesised that viruses can even hijack immune cells to produce reactive oxygen species that reduce glutathione stocks. I don’t think leaky gut helps our situation much either by dumping large quantities of bacteria and lipopolysaccharides into our bloodstream. The poor immune system and detox systems just end up with even more to deal with. Nitric oxide is another factor to touch on. This is a signalling chemical but also a free radical. Some research has found high concentrations of this substance in ME/CFS blood and it acts as a vasodilator leading to low blood pressure. Muscarinic and adrenergic receptor auto-antibodies were found in approximately 30% of patients and acetylcholine which regulates NO seems to be disturbed. Is hard to imagine that this is insignificant.
All the best.
Thanks for adding ideas :-).
“I have known 2 unfortunate people with TBI and observed a significant overlap between their symptoms and my own. ”
I try to follow the revalidation concept of TBI patients by trying to break up basic skills and relearn them such as TBI patients would be learned. I believe it is a meaningful part of my slow but continuous improvement. It is part of the “reduce energy demands on brain and body” department. Unfortunately it’s DIY as the medical profession does see no need to offer it to me…
I can imagine TBI patients develop partial mitochondria issues: part of their old “procedures” and learned knowledge and skills is destroyed by local brain damage. That makes a significant amount of brain cells to work very hard to compensate for it. That makes developing partial mitochondria problems likely IMO.
Likewise, in ME the energy dysfunctions make neural networks (learned skills) not adapted to our current brain. Trying to think and act as if we still have our old body and brain is wasting effort. Relearning it to fit our new brain and body can increase efficiency, giving mitochondria a rest. Better pacing by relearning basic skills.
“Unlike patients with TBI or post viral fatigue (often treated interchangeably as CFS but patients typically recover within 2 years), ME/CFS patients do not often achieve true recovery. Even if the brain or mitochondria are damaged in an initial “hit and run”, then surely over time the situation would improve?”
Many ME patients have ongoing underlying problems like a leaky gut (in part exacerbated by ME itself so forming a vicious circle). ME patients may often have had stronger vulnerability for underlying problems and them to be reinforced by ME itself.
So part of my ongoing “health increasing protocol” is to try and find and reduce underlying “costs” in a way that causes low side effects.
Another part is to try and find ways to increase the “self restorative / self healing” part able to get more healthy people out of this vicious circle. People can have equal health prior to a hit and run but have different “self healing” abilities. Before a strong hit, this difference remains unvisible. After a strong hit it can mean the difference between post viral fatigue and ME.
I’ll expand the ideas I wrote in my comment to Brain Thompson (bottom of the comments) with your ideas. Study patients recovering from TBI, post viral fatigue, whiplash, surgery… and study patients having ME but no FM and patients having FM but no ME and you might get a better idea of what is needed to strengthen this self healing mechanism to better be able to break the vicious circle even in current ME/FM patients.
BTW herpes type viruses like EBV are confirmed to let the cells they are in produce more ROS under certain circumstances.
I’ve always felt we were a fantastic opportunity for research, to find breakthroughs not only for us, for ones that will benefit people with many other chronic diseases. Ours is so complex that understanding of our illness will lead to understanding of basic physiological processes not yet discovered.
I’ve been taking NT Factor powder and Swanson Vitamins ATP capsules for probably 4 years. I really can’t tell
for sure whether they help. I can’t figure out whether ingesting
them actually helps produce energy or not. I’ll stop taking them for awhile then think ‘maybe’ they were helping. So then I begin taking them again. Does anyone know whether oral
ATP actually gets into cells? I get frustrated with spending the $$ on so many supplements but then think
maybe because I take so many it’s keeping me from being totally bedridden or fraught with colds or flu, which now I rarely get.
This maybe why the ketogenic diet helps some of us . I’m sure those researchers would be interested . In ketosis you force fatty acid utilisation .
My hopes are that we find CFS is mainly caused by mito dysfunction then over the next few years as genetic testing becomes cheaper and treatments are made for genetic issues we can treat the root of each persons CFS.
bold statement saying treatments for genetic issues will be available in the next few years. It seems nobody has any idea how to treat genetic mito issues. doesn’t seem close to a treatment, which is why I am praying that Ron Davis is correct about the metabolic trap. He can treat that.
Having been in the Drs. LIGHTS study in Utah on Mitrochondria and genetics, I’m going to muddy the waters even more with this comment.
My sister and I both have inherited genetics that affect mitrochondria function. The level of function could be measured with both of us. We had at least 50% of the same genes. However, the function of my sisters cells and my cells were different. She was functioning much better than me. I fell into dysfunction and into lowest 20% of function of all patients in study. At the time of draw, I was very tired and had traveled 9 hours to get to clinic for testing the day before. My sister lives there. I have many more autoimmune issues than my sis and different autonomic issues than she does. Though we both have autonomic issues. The docs felt she was probably more rested than me at draw, since we have similar genetics that could cause mitrochondrial issues. If the genes have other compensations and supporters, the overall functioning isn’t compromised. (I hope I understood what I was told correctly.) If my sis had been overstressed or overtaxed she could have manifested similar mitrochondrial dysfunctions.
Issie
As for vitiligo, (which I also have)……it could be activated Peroxide for a reason. I have fungal, Lyme and other pathogens that this could be trying to tame. There also could be amino acid imbalances. Though I haven’t found adding that amino to make a difference. It’s still progressing.
Issie
part 1
When trying to get one of my many daily restorative naps I could catch rest nor sleep. The things I just wrote kept nagging. The introduced concept was too drastic. Medical science seems to be absolutely sure the mitochondria are a main producer of oxidative stress, yet I proposed the near opposite. Sure, some researchers will have conducted experiments on whole cells and have assumed the mitochondria must be the ones responsible for the observed produced oxidative stress. But not all research will just have assumed this.
One of the things I try to do is to combine “mainstream medical science” with our “impossible disease” defying established medical science. I don’t believe medical science has it’s basics wrong. It is merely incomplete at border cases, making it seem as if though our disease is impossible. Unfortunately our disease seems to thrive at plenty of border cases combined. OR, as Cort mentions it, understanding our disease will push the borders of medical science.
So I had the problem that my hypothesis was at opposites with established experimental medical science. But as with much else in this disease I wondered whether it was not another case of seemingly opposite, due to lack of one key element in the idea.
It seems to be so. Part of the problem bugging me for a long time was how that hydrogen peroxide could be considered a signalling molecule, signalling mitochondria to produce hydrogen peroxide… That is a big kind of an unstable process. Also, how does a “bit” of hydrogen peroxide entering the cell gets through the massive amount of hydrogen peroxide the mitochondria itself produces? It kinda “gets lost in translation”. As long as this would be a self-resolving process and as long as it wouldn’t damage the mitochondria badly then it wouldn’t be such a disaster. But it is often portrait as disastrous to the mitochondria.
So I made a leap in thoughts: what if the mitochondria were doing both? Producing massive amounts of oxidative stress AND produce massive amounts of protons to shield it against the oxidative stress it produces itself? At first, the idea sounds kind of insane. Why would the mitochondria do so? Even if so, how could the oxidative stress “escape” the mitochondria and enter the rest of the cell and even get outside of the cells? If the mitochondria would both produce massive amounts of oxidative stress and protons then surely that would be one volatile reaction of both “exploding” to cancel each other out, even inside the mitochondria. Then this oxidative stress should get through the “shield” of “anti-oxidative stress” (protons, H+) surrounding the mitochondria without being cancelled out. Sure it would explain utterly excessive energy consumption with very few results. But it would be TOO costly and damaging. It would more resemble self destruction within minutes to hours then inhibition to outlast the danger for a long time.
Then it came to me: the mitochondria produce mainly (near exclusively) hydrogen peroxide as oxidative stress. And they are supposed to produce plenty of it. Now hydrogen peroxide is an oxidant that is remarkably stable. Some researchers even go that far to say that it isn’t really an oxidant as it is too stable. Things like metal oxides are far far far more reactive. But other researchers respond by saying that near all very reactive oxides are produced from this hydrogen peroxide and as such hydrogen peroxide is the mother of most oxidative stress.
Now hydrogen peroxide being that stable means it can “live” fairly longtime in an environment full of protons (H+). That means oxidant hydrogen peroxide, the main oxidant produced by the mitochondria, isn’t going to react with protons at a high rate neither in the mitochondria producing these protons nor when passing the protective shield of protons surrounding the mitochondria.
So this idea of the mitochondria producing a protective shield of protons around themselves (and also inside themselves as the protons are produced their so the concentration of protons in them must be high too) is perfectly compatible with the idea of mitochondria producing plenty of ROS.
In fact, when mitochondria are for some reason enticed to produced so much ROS, then they IMO !MUST! produce an abundance of protons so that the concentration of protons both inside themselves and surrounding their membrane is a lot higher then normal. And it seem that one effective way to achieve both things, plenty of ROS produced and self-shielding against it, is done by blocking complex V. It feels so much a deliberate choice to me now!
Doing so would be a really good cell defense against plenty of pathogens inside the cells ranging from viruses, small bacteria (like borellia) or rogue proteins. It would create an abundance of pathogen attacking oxidative stress that could be higher inside the cell then outside it. And the mitochondria would self protect really well against it (as long as one does not try to “force” once ATP production up or “unlock” the situation). I always saw this “mitochondria turning away from producing ATP to hydrogen peroxide” thing as a way to produce oxidative stress to dump outside the cells to destroy pathogens near the outside border of the cells. Turns out it resembles a very smart immune system !inside! the cells itself.
It seems to be one big machine trying to attack things entering the cells themselves. Like viruses from a big flu. Or some small subset of bacteria that can enter the cells and live and thrive there and hide from the immune system (think Borrelia with Lyme disease and its symptoms very much resembling to being near indistinguishable from ME). Think also nasty EBV that can’t get thrown out of our body by normal immune cells. Now flooding the inside of cells with massive oxidative stress does seem at least making a somewhat better chance at killing off not firmly entrenched EBV virus. Even if that doesn’t work out, it can try and destroy the viral particles that even a dormant EBV virus creates and tries to spill into the blood stream.
As to mold? Maybe it tries to fight off spores trying to enter the cell?
This defense system would also act to bacteria (or parasites or mold?…) trying to eat their way through the cell wall. It resembles the way leaves defend themselves against insects (or mammals like deer) trying to eat them. When leave get eaten the plants do two things: produce poisons that deter the animal from eating more of their leaves and produce chemicals to warn other plants to do the same. That is one reason why deer go and eat against the wind.
Now that resembles a lot what cells do under this idea. When getting “bitten” by pathogens they fill themselves up with poison (high concentrated oxidative stress). When the pathogen succeeds biting a whole in their cell wall the pathogen get flushed by highly concentrated toxic stuff. And the highly concentrated toxic stuff they produce, hydrogen peroxide, acts like a messenger to warn other cells just like what happens in the plant example.
Such thing happens best and would prevent the “runaway problem” of increased hydrogen peroxide enticing other cells to produce more hydrogen peroxide and they in turn enticing the original cells to produce more of it… if these cells don’t produce it to release it in the bloodstream. If they just (mainly) would release it when a pathogen bites through the cell wall, then this defense mechanism gets its so important ability to self limit and self clear its activation once the threat is over.
Having the mitochondria produce peroxide to pump it anyways in the bloodstream and having it increase other mitochondria to produce more of it seems unstable as hell as so easy to get out of control. I know ME is like the definition of out of control, but such system seems too uncontrollable to have many healthy people left. But a system stopping itself in its tracts if no more cell wall gets breached? Makes perfectly sense from a system design point of view.
part 2
To get back to the question: but if hydrogen peroxide is so stable how can it act a such a toxin to pathogens? Well, hydrogen peroxide is the main source of far far more reactive super oxides that are very destructive. And a pathogen does not seem to be needed to be attacked and destroyed in seconds, so having super oxide after super oxide attacking it all day long will do to the pathogen as it did to my patch of skin as I described it in my first comment: weaken and dissolve it over a day or more. Probably works well enough in many cases.
That also gets us to another question: but if hydrogen peroxide reacts so few with these protons, how can a “proton shield” protect the mitochondria against oxidative stress? Well, if hydrogen peroxide is so (relatively) stable then the mitochondria do not need to fear it. But they do need to fear the super oxides formed slowly by the hydrogen peroxide. Most of these will be formed in the liquid inside the cells but outside the mitochondria. It these destructive molecules get near the mitochondria and their proton shield, they react away at lighting speed with the mitochondria. Danger neutralized. If these super oxides form within the mitochondria: they will be canceled by the high proton concentration inside the mitochondria very quickly too. Remember: in order to get many protons from the mitochondria to the membrane it “helps” to produce plenty in the mitochondria. And producing plenty of them in the mitochondria makes it a hyper metabolic disease as some researchers call ME. Having fewer (at exercise) of those protons turn to ATP makes it a hypo metabolic disease. That ticks another astounding self-contradiction of ME.
It also explains why a “mere” drop of 25% complex V activity causes complex I and others to go “gangbusters” as Cort calls it: much of the produced protons are “eaten away” when they exert their role of protecting the mitochondria against the plenty of super oxides nearing their borders. With so much super oxides nearing their borders because the mitochondria themselves produces so much of its much more stable precursor hydrogen peroxide just to defend the cells against pathogens.
It also would help explain why (calcium) ion channels are so often mentioned with a disease like ME. In this hypothesis they would NOT be defective. Having different calcium and other ion settings is *required* in this model in order to be able to “maintain” a proper big sized proton shield. Having a bigger amount of protons (for defending against oxidative radicals and super oxides) builds up extra charge. Having these protons remaining at a stable position around the mitochondria requires a different (compared to healthy people) amount of ions both inside the mitochondria and inside the cells. We may not have calcium ion problems, but adaptation to this defensive mechanism. The adaptation may not be perfect but trying to get better values with drugs may be difficult, sensitive and tricky.
Now this, if I say so myself, innovative idea has potential to explain so much unaccounted and weird things going on in ME just by thinking it over a few hours. Now that oozes potential to be explored further ot see where it could get us.
Just popping into my mind: over exertion or plain exercise would “demand” more ATP production “eating away” part of the proton shield, making our cells defenses against either the oxidative stress, the pathogen or both falter. That would help explain why a few weeks of too much exertion creates patches of skin “eaten away by the immune system”. Exercise induced exhaustion and disease seems to be almost a natural outcome of this model :-).
It would also help explain why researchers see a *delayed* increase in oxidative stress after exercise. It would eat through the “second protective proton shield” located near the inner cell wall creating cell wall breaches leaking out oxidative stress in a delayed fashion only after the oxidative stress slowly eats through the cell wall. See explanation of this “second proton shield” below as I added this when re-reading my comment.
Remaining question under this hypothesis:
* Is the threat over? OR in other words is the system getting stuck?
=> If the system would require cell walls to rupture the the question remains: what are the protections the inner cell wall has against these very reactive oxidative stress components?
It should, as the supposed high amount of oxidative damage could weaken the cell walls and perpetuate the problem. One could call it the cause of our disease being stuck and in a vicious circle in this model. But I believe the inner cell wall *must* have a protective system all by itself. If not so, the faintest immune challenge would get healthy people stuck in this lock in.
The “calcium ion channel problems” may come to the rescue here too. If the charge of calcium (and potassium and other) ions is “unbalanced” compared to the charges outside the cells “due to defect ion channels” then a charge imbalance occurs. If “done well”, that will attract some of the protons produced by the mitochondria and let them stick to the inner cell walls. That creates a second “proton shield”. It probably would be weaker then the one surrounding the mitochondria membrane but it would offer some protection. Maybe that could make the difference between getting ME and not getting ME: the weakest of the two “proton shields” gets breached after too strong or too long an assault on the shield. That would cause a very strong or a long lasting attack to drag people into ME. Here again “correcting” ion channel problems may be quite a tricky thing to do.
For the people saying “but if the mitochondria are surrounded by protons and the inner cell wall is surround by protons then they must be everywhere and then all damaging oxidative stress is canceled making the system worthless” I answer: charges have a *very* strong tendency to stick to surfaces with a voltage difference on both sides of the surface. And protons are charged particles so they stick to surfaces. So, if voltage differences over membranes are set correctly, it is entirely possible to have plenty of protons on both surfaces and only small amounts in between them. These small amount may be enough to transport enough charged protons from the mitochondria to the inner cell wall, creating this second shield.
I couldn’t help but be reminded of something in my one multi-year experience with ME/CFS. This quote: “Some of the electrons involved in the energy production process inevitably leak out of the mitochondria, create reactive oxygen species (ROS), and attempt to tear apart any cell they come into contact with”. I had been feeling rundown for quite some time – less up for going to the gym when I had been all into it. Less energy at work, etc. Then in 2012 I was hospitalized for a splenic infarct as well as an infarct in one kidney. The admitting doctor in the ER told me “I may write a paper on you; your spleen is tearing up all of your red blood cells”. I didn’t put it together until years later, once I had a diagnosis (we know how long that can take). It wasn’t my spleen causing the blood clot; it was a collection of the “garbage” in my blood that likely caused those clots. That same ER doc said that he’d never seen that many broken cells in anybody’s blood before. Anyway, ME/CFS may or may not have been the cause of those infarcts, but I believe it was the culprit. I never really recovered from that hospital episode, but it did set me on a search to find out what was wrong. Just thought this might ring a bell for someone else out there. Has anyone else had blood clots or an infarct since they’ve had ME/CFS
Part of this very new “protective proton shield” hypothesis I’ve been spamming this blog with concerns RBC a lot. In this hypothesis it are the mitochondria that both produce the hydrogen peroxide and get them and likely the cell walls of the cells they live in some protective shield. Now RBC lack mitochondria as they need to be small enough to work well.
No mitochondria, no protective “proton shield”. So they are basically sitting ducks. Or almost. Long I believed that the RBC’s hemoglobin was the last line of defense against oxidative stress if all other oxidative defenses like glutathion failed. That impairs breathing, but helps protecting the body from oxidative stress. Damn if you do, damn if you don’t…
But now I tend to believe its is only the last line of defense for cells without mitochondria. And that mitochondria may not be the most vulnerable part in ME, but the (inner) cell walls.
We’ve read about inflexible RBC (due to oxidative stress damage) having a hard time to pass through capillaries a lot on HR recently. But your “I may write a paper on you; your spleen is tearing up all of your red blood cells” may be telling: when glutathione and other oxidative stress defenses are down, RBC are among the weakest sitting ducks it seems. Happen to had a very rough time breathing around that period (as offering / temporarily sacrificing their hemoglobin by binding it to peroxide seem to be their last defense, making oxygen uptake very poor)?
It may have been the oxidative stress ripping apart your RBC. It happening in the spleen may be the consequence of the spleen seemingly “collecting (and maybe recycling)” RBC so there would end up very high amounts of broken RBC in your spleen.
(Maybe too far off your question, but lactic acid also produces protons but also other negatively charged particles (making estimating its impact difficult) so ME and too much oxidative stress could mess with lactic acid in the body both as a sign of overexertion or as a source of energy; RBC use anaerobe energy production producing plenty of pyruvate and maybe plenty of lactic acid).
part 3
Do I have some proof of what I wrote in part 1 and 2? NO!
But I did found quite a nice thing “summing it up”.
When I tried to search how well hydrogen peroxide could transport through the mitochondria wall in order to estimate how likely it would be the described system could work, I entered “peroxide mitochondria membrane” in my favorite search machine Duckduckgo (as it doesn’t personalize results and makes them more repeatable).
First hit:
ncbi.nlm.nih.gov/pmc/articles/PMC3278611/
with title “Role of mitochondrial dysfunction in hydrogen peroxide-induced apoptosis of intestinal epithelial cells”
The short description in the search engine said:
“We observed that hydrogen peroxide could result in the collapse of mitochondrial membrane potential, if we related it with the release of cytochrome c, we may get the conclusion that hydrogen peroxide caused the release of cytochrome c from mitochondria to cytosol followed by the increased permeability mitochondrial membrane and the opening of …”
Now a collapse of the mitochondria membrane potential by adding (plenty?) of external hydrogen? Now how could such membrane potential collapse? Or in other words how could the voltage difference disappear? The most simple way I know as an engineer (and engineers have capacitor as a standard physics topics that nearly perfectly describes this kind of charged membranes with charges and potential differences across them) is to simply remove the charges.
How? One could go through the complex mechanism of altering the entire mitochondria chemistry to the point they produce no single proton (unlikely IMO) or far more likely the massive amounts of !external! hydrogen peroxide causes more reactive oxidative stress species quickly enough to “eat away” all protons on the outer side of the membrane (result: no protons equals no mitochondrial membrane potential, simple as that). Sounds VERY much as what I described in the previous comments.
And the title resembling: “death of intestinal cells by massive amounts of ROS” sounds a lot like the skin observation I described above. With explaining why exertion could increase oxidative stress imbalance in the exercising cells.
Why would that cause my skin to be eaten by my immune system or by hydrogen peroxide? Well, I dislike the explanation myself. Too much exertion of my brain and muscle cells cause these cells to produce more ATP and hence loose part of their inner cell “protective proton shield”. That allowed the high amounts of hydrogen peroxide and the super oxides it produces to slowly punch more and more holes through these cell walls and let them leak hydrogen peroxide into the blood stream increasing oxidative stress throughout the body.
That in term resembles the “high amounts of external hydrogen peroxide” applied to other cells like my patch of skin eaten away. These amounts seem to be high enough to slowly eat through my skin and dissolve part of it. That also sounds a likely cause of my gut hurting a lot more now.
Note that gut problems often cause necrotic parts of the bowel (“induced intestinal cell apoptosis”). Ouch, I dislike the idea of sections of necrotic bowel as that is very difficult to restore. But it also links to why infection, exercise (see recent ME studies), stress… all can cause gut inflammation. This also seem to flow “naturally” out of the proposed hypothesis.
Now why do I dislike this description even more? If the hydrogen peroxide that leaked mainly out of my brain cells with breached cell walls (as the exhaustion of “choice” was mainly mental) creates such ugly wounds on my skin as a secondary effect, then how ugly must the damage look to my brain cells? Gulp. Would that be the “mild” brain inflammation some researchers find? Kinda puts “mild” into perspective :-(.
Note: still need to read the paper in depth.
Please bring on the comments, I feel like being on a roll with being “creative” now :-).
I can tell you what your brain looks like and so can a special MRI called a Neuroquant. It measures your brain and shows atrophy and swellings. It can let you know if something is too large or has shrunk. It’s done to help DX CIRS. Here is an article on findings.
https://www.ncbi.nlm.nih.gov/m/pubmed/24946038/
With my having had CIRS, I’ve had several of these through my treatment period. We keep a check on my inflammation factors.
Another thing to also consider……they are starting to find that inflammation plays a huge role in Alzheimer’s disease. Which also can cause brain shrinkage. Lots of info and new ideas on treating brain inflammation, which can also treat body wide inflammation. Hopefully protecting or reversing damage already done.
Sooooooo, none of this is good for the brain.
Issie
When thinking further on the hypothesis, it helps me understand why calcium is called to play an essential role in programmed cell death.
From ncbi.nlm.nih.gov/pmc/articles/PMC4556774/ with title “Calcium and ROS: A mutual interplay”:
“Calcium is an important second messenger involved in intra- and extracellular signaling cascades and plays an essential role in cell life and death decisions.”
The paper describes plenty of hard to understand biochemical mechanisms how calcium ions affect cell life and death.
In my “proton shield” hypothesis this may be (over)simplified to:
Without the proper amount of calcium charges in the cell or in cell sub units like mitochondria one cannot have the correct voltage potential across the cell or cell compartment membrane. Without the correct voltage potential one cannot have the correct amount of protons (H+) on the other side of the membrane.
If this voltage potential setting is such that it decreases this proton layer to (near) non existent then there simply is no proton shield to protect against the very reactive oxidative stress species produced by hydrogen peroxide. No such protection and the cell or cell compartment wall gets eaten away very quickly. Result: cell death.
In true programmed cell death there is a “nice, correct” sequence of what parts are destroyed first in order to make cleaning the mess up easier and produce far less toxic waste. Note that programmed cell death is essential to good health so it’s not all bad.
When the “proton shield defenses” are overrun however things get far more messy: unclean but probably even quicker cell death.
In that model, ME is an inhibited auto apoptosis disease. Without this inhibition, an auto apoptosis disease would be worse then an auto inflammatory disease and an auto immune disease !combined!. In fact it would be nothing less then sort of an auto immunity disease to *all* cell types the body has.
In this model it is no wonder that there is no main primary auto-immunity marker in ME (but their can be many secondary ones as observed). It is the massive production of ROS that is the auto-immunity marker itself that works against all cell types. This massive production is only masked by an equally massive production of excessive amounts of protective protons for continuously replacing the lost protons in the fast turn over proton shield. That plus the inhibition aspect of ME make it difficult to “appreciate” the true extent of massive body wide ROS production and this hard to measure massive ROS production as a marker for an (inhibited) auto apoptosis disease. One most easily can measure just the resulting amounts of present ROS, which are kept somewhat in check by both mechanisms.
Undoing this inhibition (in a bad case of ME), like some desire, would result IMO in a body wide auto apoptosis with speed and strength that would make full blown sepsis pale. We are talking about a very nasty looking death in all organs within hours in bad cases of ME IMO.
Even in the (likely minority amount of) cases where there is no longer a ME “promoting” problem left, just blocking this inhibition without first tempering down this auto apoptosis aspect a lot would result in auto destruction in hours. I have a hard time seeing a drug able to resolve both the auto apoptosis aspect and the inhibition aspect simultaneously within hours after a single drug infusion.
May I ask which criteria were used to create the cohort please?
I don’t know. We’ll find out when the paper comes out which I believe will be soon.
I’m glad to see these studies. Mitochondrial disease is tricky for exactly the reasons mentioned, and also due to a high degree of heteroplasty leading to variable concentrations of dysfunctional organelles in different tissues, and sometimes even the same tissue. For example, a muscle biopsy of the leg may show little dysfunction but a biopsy of the heart could be severely affected. Plasma is easy to work with, but it may not reflect the full severity of the issue. A good place to look is in any tissue that cannot stop working but unfortunately it’s not practical to biopsy these areas.
Interestingly, recent work by mitochondrial pioneer Dr Doug Wallace showed that an induced mitochondrial defect in mice that resulted in a very similar pathology to what is described here resulted in behavioral changes that mimicked CFS symptoms. The mice exhibited normal energetics and normal behavior until stressed, at which point the energetic deficit became measurable. I’ve been grinding my teeth ever since I heard that and hoping someone would test for it!
CFS research focusing on Mito has always been fraught with core misunderstandings of the disease, leading to improper conclusions. It’s nice to see someone getting it right.
More or less proven by response between my sis and I. Funny thing is her fatigue has always seemed worse than mine, yet my energy factors (mitrochondria) was at a super low of energy or functioning.
Issie
I hate to hear these findings because if this is our issue it seems nobody has any idea how to fix it and we’re pretty much screwed
A few thoughts after reading Cort’s article and dejurgen’s many musings:
1) Thanks for another great article, Cort!
2) dejurgen, your many thoughts could generate a great discussion over in Phoenix Rising – can you come over there and share, please?
3) Some of this needs to be looked at in context with Neil MCGregor’s July 4 article, which points to low hypoxanthine, issues with glycolysis and acetylation.
4) My complex I is not overworking – it’s been tested at 30-50% of normal.
5) Complexes II and IV have been tested at 370% of normal, generating lots of superoxide radicals that create peroxynitrites and hydrogen peroxide (confirmed by testing) that damage mito membranes and impair complex I, a vicious circle. Have not had complex V tested, but plan to.
6) This vicious cycle has positively responded to folate, B12, vitamin C and other mito friendly nutrients and phospholipids in the form of NT Factor and IV phosphatidyl choline. Tests revealed lower lipid peroxides, lower peroxynitrites, more normal mito function and lower 8OH-dG, a marker of DNA damaged, with greatly improved energy. NMN/NAD+ have helped too.
7) I’m having a hard time folloowing the inconsistencies between what Davis, McGregor, Fisher, and the others have found about mitochondria. There’s no ptpblem with mitos, yes there is, it’s complex V, it’s complex I, it’s glycolysis, it’s beta oxidation, etc. Could be there are different subcategories of patients…
It is clear that something is going on with mitochondria and more patients should have access to testing and tools to figure out more about what is going on.
Would like to see a conference around this topic…
Learner 1 – how and where did you get the testing. The only MD I have found with any interest or knowledge on CFS/ME was my eye surgeon.
2): Sorry, no. I still can’t handle the complexity of strong interaction on different sites and remember where I wrote what.
Cort generates the best synthesis of past research for me to work with, reducing the complexity of my “task” a lot. So here is my base. You can reference others to my ideas and this site though :-).
3)
“low hypoxanthine”
Xanthine oxidase is influenced by both hypoxia and ischemia, not totally strange to ME. That affects hypoxanthine content. ” researchgate.net/publication/14438940_Regulation_of_xanthine_dehydrogenase_and_xanthine_oxidase_activity_by_hypoxia: “Regulation of xanthine dehydrogenase and xanthine oxidase activity by hypoxia”
physiology.org/doi/abs/10.1152/ajpheart.1988.255.6.h1269:
“Role of xanthine oxidase and granulocytes in ischemia-reperfusion injury”
“, issues with glycolysis”
In healthy people the rate of glycolysis (pyruvate production) is far far greater (800 times?) then the rate the Krebbs cycle can consume pyruvate.
So therefore partial inhibition of glycolysis needs to utterly destroy anaerobic functioning (safe for creatine reserves) before significantly reducing the speed of the Krebbs cycle.
AFAIK McGregor did find a decrease, but did not presented numbers or an indication numbers are that massive.
IMO a significant reduction of glycolysis is good to slow down anaerobic metabolism (conserving energy, improving inhibition) and a simultaneous significant inhibition of glycolysis in the part exclusive to glycolysis (not common with the PPP) and an activation in the Penthose Phosphate Pathway is the best way to displace resources from producing NADH/ATP towards NADPH/recycling glutathione. The latter is very important in a situation of strong oxidative stress.
As there are already 200 proteines (mainly involving energy metabolism) found that have this “serine-switch” effect based on amount of oxidative stress, doing tests using ME plasma versus using a clean environment may include more inhibitory chemicals. But both seem to inhibit energy production under heavy oxidative stress.
” and acetylation.”
Still alchemy to me
3)
The most important part of the McGregor study IMO may be his demonstration that cells are glucose starved during part of the night.
That makes maintaining the hyper-metabolic part of ME, producing plenty of oxydative stress to fight off pathogens and producing a good “H+ proton shield” defending the cell itself against it, near impossible in my hypothesis. During the night, both defense against potential pathogens and cell wall defense against the high amounts of oxidative stress suffer causing massive damage before morning comes. That in turn makes nights deplete our health and energy levels below the level we had when going to bed.
ME patients reduced blood volumes, reduced blood flow and contracted blood vessels makes blood flow to part of the body during part of the day very problematic destroying our defenses much more then what healthy people experience IMO.
“4) My complex I is not overworking – it’s been tested at 30-50% of normal.
5) Complexes II and IV have been tested at 370% of normal…”
That may indicate the “In Vivo” (aditional, on top of Fischers observation) impact of oxidative stress from outside the cell impacting the Krebbs Cycle.
In previous searches (regarding hydrogen peroxide applied to full cells externally), I did found that alfa-ketoregulate, aconitase and succinic dehydrogenase are all enzymes that are impacted (inhibited) due to strong oxidative stress.
Alfa-ketoregulate and IMO succinic dehydrogenase are inhibited relatively linear with increasing oxidative stress. Aconitase stay functioning close to 100% until very high amounts of oxidative stress and then drops functionality like a falling stone.
See en.wikipedia.org/wiki/File:Citric_acid_cycle_with_aconitate_2.svg for the interaction point of these enzymes.
Now from en.wikipedia.org/wiki/Electron_transport_chain see the “text-schematic” above the flashing picture / “movie”
It tells that “Q” can com fro *either* complex 1 or complex 2.
Now that is not clear enough. Let us look at biologywise.com/electron-transport-chain-steps-explained-with-diagram, first schematic.
It tells that Complex 1 mainly uses NADH as both an energy source and a source of protons (H+)
It tells that complex 2 mainly uses succinate to fumarate as a source of energy to produce quinones that are further “processed” by complexes 3 and 4.
Now it seems you have much higher succinate to fumarate conversion then NADH production. Several options exist. Most involve that you don’t use “full Krebbs cycle” but more the “Krebbs roundabout” with hop on hop off before a cycle is full:
Somehow you don’t use any NADH producing steps; unlikely IMO as it seems difficult to get enough resources like succinyl coa or succinate and be able to dump fumarate or malate quick enough.
So you still seem to produce some NADH, but far less then succinate and fumarate (30-50% versus 370%)
How is this possible?
IMO both the alfa ketoregulate to succinyl coa and the malate to oxolaacetate steps have variant enzymes that produce NADPH rather then NADH. Most likely they don’t produce full 100% of their conversions NADPH and still some NADH. With a near 1 to 10 ratio of NADH versus quinone production, even at roughly 90% NADPH production versus NADH production of both steps that would make that the D-isocitrate to alfa-ketoregulate step is near completely running empty.
That in turn would point to “pretty extreme oxidative stress” surrounding the cells as it would indicate that aconitase is near completely inhibited and with it still functional at very high oxidative stress but dropping like a stone at extreme oxidative stress… that tells a story.
Also needing to produce about 90% NADPH in steps normally producing near only NADH tells how humongous amounts of NADPH are consumed (probably for both immune cells using them to create an oxidative burst and for glutathion recycling for cleaning up the massive oxidative waste) tells a story if wright. (Note that this is my older “energy production massively switches to NADPH production story seeming to fit your measurement data).
Now what sources of energy can then be used:
1) glutamate as a source of alfa-ketoregulate (and this conversion also IMO able to produce both NADPH and /or NADH).
2) source of protein “equal to” glutamate
3) sources of succinyl coa; few exist for humans; from wikipedia.org/wiki/Succinyl-CoA:
“With B12 as an enzymatic cofactor, it is also synthesized from propionyl CoA, the odd-numbered fatty acid, which cannot undergo beta-oxidation.”
=> basically eating diary if you can tolerate or (grass fed?) eating ruminant meat (goat, cow…). Note that the odd-numbered fatty acid is about less then 10% of energy content on average.
From wikipedia.org/wiki/Propionyl-CoA:
“Propionyl-CoA is not only produced from the oxidation of odd-chain fatty acids, but also by the oxidation of amino acids including methionine, valine, isoleucine, and threonine.”
“Cholesterol oxidation, which forms bile acids, also forms propionyl-CoA as a side product.”
4) succinic acid wikipedia.org/wiki/Succinic_acid:
“As a food additive and dietary supplement, succinic acid is generally recognized as safe by the U.S. Food and Drug Administration.[19] Succinic acid is used primarily as an acidity regulator[20] in the food and beverage industry.”
=> be aware it may have side effects when supplementing in ME!
Supplementing GABA: “GABA is then metabolized by GABA transaminase to succinic semialdehyde. Finally, succinic semialdehyde is oxidized by succinic semialdehyde dehydrogenase (SSADH) to form succinate, re-entering the TCA cycle”
Note 1: GABA is a know food supplement that helps many but is a disaster for a minority of patients with hard to revert loss of health.
Note 2: Glutamate: same as 1
Note 3: Some “chronique fatigue patients” return to near health with regular strong vitamine B12 injections and option 3) should “rip through” vit B12 levels if the body would chose this route; requires odd chain fatty acids to work!
Note 4: many ME patients are said to have really low cholesterol levels, might be “consumed” to form propionyl coa
“4) My complex I is not overworking – it’s been tested at 30-50% of normal.
5) Complexes II and IV have been tested at 370%”
Now that still should generate far more protons then when all complexes work at 100%, still leaving the “proton shield idea” as an option. Only complex I would falter as well when put into ME plasma with excessive amounts of oxidative stress (versus ideal experiments of Fischer). See above comment.
Likely, near inhibiting citrate conversion however would strongly reduce the overall production capacity of the KRebbs cycle / roundabout as fuel for the active part described in previous comment would be scarce.
Also, on top of decreased “total Krebbs throughput” in real plasma versus ideal circumstances, excessive amounts of oxidative stress may produce up to or around 90% of NADPH versus NADH! that would not yield 90% loss off ATP production in the (used part) of the Krebbs cycle, but (very rough guess, need to find info back with decent numbers) about 60% loss as quinones produce ATP as well.
That 60% loss would be on top of:
* reduced total output due to “correct fuel” shortage
* proton / H+ loss due to “recombination with oxidative stress” in its functioning as oxidative stress shield
* loss of electrons in order to produce plenty of hydrogen peroxide for cell defense
=> That sounds much more like the energy loss I experience in daily life!
6)
Vit B12: see above
Vit C, good anti oxidant cleaning up oxidative stress; may be hard to get orally in sufficient quantities to make a dent.
May work in “normal” quantities due to anti viral properties reducing “threat” to the body or, when applied well, due to controlling amount of gut bacteria.
Rest: haven’t looked into it yet.
“7) I’m having a hard time folloowing the inconsistencies between what Davis, McGregor, Fisher, and the others have found about mitochondria. There’s no ptpblem with mitos, yes there is,”
Fischer studies “well fed” cells that are not subject to external oxidative stress (as far as we know). Oxidative stress may reduce the amount of energy resources available to the mitochondria at great throughput. Poor blood volume and flow may hamper part of the body from getting good nutrient access.
That all may make the hyper-metabolic part (producing plenty of protons, NADPH for glutathion recycling) impossible for these cells leaving them far more vulnerable to damage.
Note also: cell type and how long a patient is ill may make a very big difference in how much mito damage has accumulated.
Mito damage not being obviously observable in all studies seems to point that it is “only” a secondary effect of this disease IMO, not the “origin”. It still can additionally deteriorate patients health over time when it accumulates IMO.
it’s complex V, it’s complex I, it’s glycolysis,
see above
it’s beta oxidation, etc.
If many patients follow your “complex profile” and my analysis of it, both glucose and most parts of fatty acids get very slowly through the aconitase step so active consumption / conversion is slowed down a lot, but mainly due to one oxidative stress affected enzyme.
I may too easily have used 50% activity and 370% activity as meaning “produce 50% of normal” and “produces 370% of normal” as someones definition of activity may differ from mine.
Still, the lack of good fuel sources for your measured values was nagging at me. As I wrongly remembered to have read that some bacteria can produce succinate while writing above comment, I decided to search for “succinate producing gut bacteria” and got several hits.
cell.com/cell-metabolism/fulltext/S1550-4131(16)30299-6 with title “Microbiota-Produced Succinate Improves Glucose Homeostasis via Intestinal Gluconeogenesis”
“The microbiota produces high amounts of succinate in response to dietary fibers”
Might that be important to some of us? Might the gut alter it’s (immune) functioning to accommodate more of them under high oxidative stress conditions…
“succinate should not accumulate in the bowel to a substantial extent (Macfarlane and Macfarlane, 2003). Interestingly, its concentration in the cecum is increased by feeding dietary fibers (Everard et al., 2014, Jakobsdottir et al., 2013), and this increase is even more significant when dietary fiber is fed in conjunction with high-fat diet (Jakobsdottir et al., 2013, Zhong et al., 2015).”
Thank you for this article, Cort!
There sure is a great opportunity for the person who figures out this disease to have fame and glory and maybe even to name the illness.
wow! great in depth work that can’t help but help!
(sidenote: if anyone interested in an article explaining use of energy by brain—such as burning of fatty acids.
“”our hypothesis, presented here (see Figure 5), β-oxidation of fatty acids is the source of acetyl-CoA for the TCA cycle. ””
“”β-oxidation of fatty acids would supply carbons for anaplerotic synthesis de novo of glutamate and glutamine, this would spare glucose for other functions.””
(had to skim and did not make it anywhere near end of article.)
wow! great in depth work that can’t help but help!
(sidenote: if anyone interested in an article explaining use of energy by brain—such as burning of fatty acids.
“”our hypothesis, presented here (see Figure 5), β-oxidation of fatty acids is the source of acetyl-CoA for the TCA cycle. ””
“”β-oxidation of fatty acids would supply carbons for anaplerotic synthesis de novo of glutamate and glutamine, this would spare glucose for other functions.””
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4026875/#!po=0.240385
(had to skim and did not make it anywhere near end of article.)
Thanks for that link. Seems important as to what mitrochondria and brain function is. Seems to think some of the low levels are compensatory. I had been told to use mitrochondrial support supplements. Some seem to help a little and others didn’t. Succinate had been suggested and I didn’t find it to do much for me. According to this article it’s old thought and not best solution.
I’ve always thought glutamate and its excitatory response potentially could be responsible for some of our symptoms with high sympathetic nervous system response with POTS. Yet it appears from this article it could also be assisting our mitrochondria. I found with myself, that I didn’t need to suppress this response completely. I seemed to do better to have a portion of over sympathetic responses. But there is a fine line as to how much.
Also do feel there is a connection with calcium channels.
Have also felt my high Hydrogen peroxide was compensatory response with vitiligo being a result of a compensation.
Will be interesting to see what time and our hypothesis brings.
Dejurgen keep it coming. Love to read your thoughts. Nothing wrong with your brain…..see what sorts out with time. (Check your PM from forum.)
Issie
Nancy Haehnel your comments interest me greatly. In 1997 following a significant crash I developed multiple pulmonary emboli and DVT. Later understanding had me thinking much the same as you that my blood had thickened in some way.
12 months ago I developed another DVT following a flu shot. I suspect the stress on my body caused a similar reaction. I have no other co- factors for risk of DVT.
Thanks for sharing your story
Dumb question but what do I do when having breathing and respiration problems. At the cellular level is it similar to what I experience frequently. Right now as I am propped up in bed using the computer. I have had a severe relapse, a simple cataract surgery has knocked me down. These breathing problems occasionally surface. Any ideas?
Interesting Linda. I too have breathing difficulties at times. They are treated as asthma and I have inhalers – quite strong ones now. The nurses/doctors are always surprised how often/much I need inhalers, even though my ‘peak flow’ readings seem quite good.
I know I DO have asthma – a history of bronchitis as a child, growing up near coal mines and developing allergies all contribute to that.
But I’ve sometimes wondered whether ME/CFS is playing a role in my breathing problems. I’ve had times when I’m in a crash when I can’t talk because it makes me breathless. I also have times when the inhalers don’t seem to make any difference and I just have to use calming meditation and some Feldencrais positions to breathe slowly and deeply.
I live in UK and have never mentioned this to any Dr as none of them understand ME/CFS enough. They mostly know it’s a real illness but also that they don’t know what to do to help. When I told my Dr what my approach was (supplements, pacing, drinking salt water, adopting a low carb diet) she said, “Well, you know more about this than I do, just don’t do anything wacky you hear about on the internet.” Basically, it’s online research (Yay! for Health Rising!), trial and error, listening to my body and following my instincts. I do worry sometimes that, though the things I’m doing/taking are helping day to day, I might be causing some long term problem in my body that I don’t understand about, but I don’t have any option really. It’s a bit scary sometimes. I’m just glad I have a wonderful husband and family and our finances are generally flexible enough to keep buying the supplements.
Thanks Cort for the encouraging and informative posts!
Cort,
In my opinion, this is one of the most discouraging pieces of research that I have come across. Because to me this suggests that we have a root problem that is incredibly difficult to fix and we are pretty much screwed if this is true.
Not necessarily,
I am not a medical researcher but If my latest ideas are close to wright, this research helps indicating that ME and FM are close sister diseases.
My “cells self protect by producing poison (concentrated oxidative stress) that gets released upon the pathogen when cell walls are breached” idea is not that far from the observation there is plenty of fibrin in the fascia. Fibrin is a protein formed when two components meet when there is clear damage at the wall that separates them.
Here that wall seems to be the cell wall with the cells holding component 1 and the fascia holding component 2. When cell walls falter both components form “hole sealing” glue to repair the wholes.
If so, then the process to the muscles is similar to and less difficult to understand of what happens to the brain (as it has no fascia and Philippe Hayward can’t offer clues there).
Good point: IF the underlying process is similar and IF both ME and FM are self resolving when taking away remaining underlying causes then studying FM patients having no ME and ME patients having no FM can learn why they get out of the vicious circle for the sister disease they don’t have. That could reveal soooo much I hope so.
I was an athlete before cfs/me hit me. I used to love that lactic acid muscle pain after a big event. What i have found since coming down with cfs is i dont seem to have sore muscles from lactic acid build up any more even when i rarely push myself. Its like anerobic glycolisis doesnt happen any more.
Is it possible we retain too much calcium phosphate during the ATP cycle?
Taking 5 drops 1% solution USP, but “tired and wired”.
Cannot take an evening dose, as inhibits sleep for a few hours.
Some increased exercise and cognitive effects, but also a feeling of fatigue in head.
Comments????