



Paul Fisher wanted to study ME/CFS ten years ago…(from https://www.latrobe.edu.au/our-work/parkinsons/from-accident-to-medical-breakthrough)
Frustrated for 10 years in his desire to study ME/CFS, Paul Fisher of LaTrobe University in Australia made the most of it when he got the chance He and his lead researcher on these studies, Daniel Missailidis, have rolled off four studies and two reviews in the last three years. They include the most sophisticated analyses of mitochondrial functioning done yet in ME/CFS and have provided a new slant on what’s possibly going on.
First, though, a quickie overview of mitochondrial energy production.
Mitochondrial Energy Production Simplified
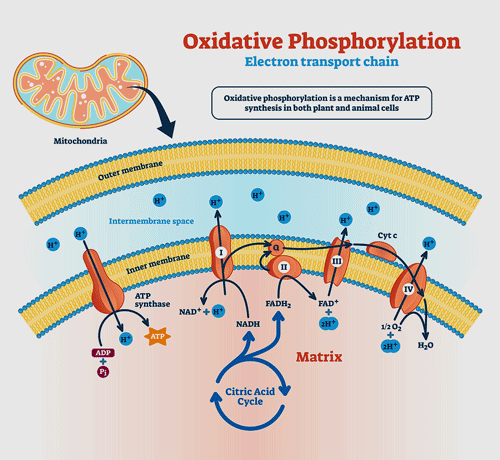
Producing ATP involves two processes that work side by side. The TCA, or Krebs, cycle provides the fuel the electron transport chain (ETC) needs to function. The ETC then uses electrons to convert ADP to the usable form of energy for the cells – ATP.
As the electrons go through Complexes I-IV in the electron transport chain, they cause protons to get pumped out into the spaces between the mitochondrial membranes. When the protons reach a certain level, Complex V is able to transform ADP to ATP. With that, the electron transport chain’s job is done: it’s produced the energy the cell needs to run on.

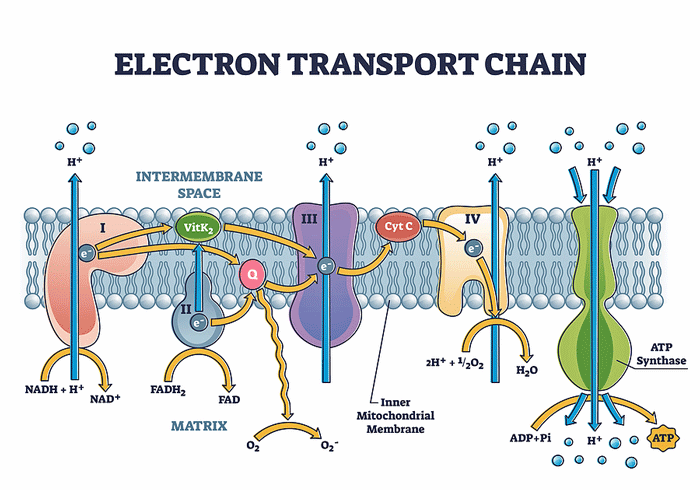
Missailidis and Fisher’s studies suggest that something has gone very wrong with Complex V in the electron transport chain.
It’s critical, then, that the electron transport chain has the fuel – NADH, and FADH2 – it needs to run on. These compounds are produced by another process entirely, which runs alongside the electron transport chain and provides it with the fuel it needs. It’s known by a couple of names: the Krebs, TCA (tricarboxylic acid cycle) or citric acid cycle.
(Hans Kreb was a German Jew who was dismissed by the Nazis in 1933 and ended up in Oxford, England on his way to a Nobel Prize and enormous acclaim. His letter describing the cycle was rejected by Nature, but his manuscript was later published elsewhere. He won the Nobel Prize for the discovery in 1953. He also discovered a couple of other cycles. :))
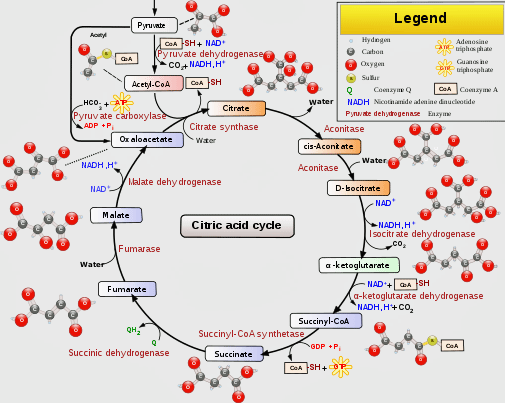
The Krebs aka TCA aka citric acid cycle provides the electron transport chain the fuel it needs to produce ATP. (From Wikimedia Commons https://upload.wikimedia.org/wikipedia/commons/thumb/0/0b/Citric_acid_cycle_with_aconitate_2.svg/512px-Citric_acid_cycle_with_aconitate_2.svg.png)
First, two compounds, NADH and FADH2 need to be “reduced”; i.e. electrons need to be added to them so that they can give them up later in the first four complexes of the electron transport chain. (Adding negatively charged electrons to a compound results in that compound being “reduced“).
Glucose, fatty acids, and amino acids can all be used by the TCA, or Krebs, cycle to produce NADH and FADH2. First, though, these substances have to be metabolized into a compound that the TCA cycle can use. For glucose and fatty acids – the two main sources of fuel – that compound is acetyl CoA.
- Glucose – glycolysis provides the mitochondria with pyruvate, which is then converted by pyruvate dehydrogenase (PDH) into acetyl CoA.
- Fatty acid – the beta-oxidation of fatty acids provides acetyl CoA.
Amino acids are different. The relevant amino acids are broken down by a variety of methods. For instance, glutamine is converted to glutamate, which is then converted to alpha-ketoglutarate – which can be used by the TCA cycle.
The Glycolysis Hypothesis
Since three plasma studies pegged a deficient glycolytic process as the main potential culprit in ME/CFS glycolysis – which uses glucose to produce acetyl CoA – it has been the main focus. (Plasma is a clear liquid that doesn’t contain red blood or other cells).

With different studies getting different results the mitochondrial field has been something of a “dogs breakfast”. Fisher, though, believes he’s found a clear and specific abnormality.
Studies using serum, however, were unable to replicate their findings. (Serum doesn’t contain cells either but it also doesn’t contain any clotting factors.) Seahorse studies have also failed to find evidence that glycolysis is inhibited in ME/CFS. Some studies have also suggested that amino acids – rather dirty fuels – are being preferentially utilized to produce energy in ME/CFS.
With virtually every study finding something wrong, but different studies pointing at different things, the quest to pin down the energy problem in ME/CFS patients’ cells has become something of a “dog’s breakfast” at this point; i.e. it’s kind of a mess.
Special Cell Line Plays Significant Role
Enter the Fisher group with a new method of studying the mitochondria they hoped would bring clarity.
In the 2020 review, “Lymphoblastoid Cell Lines as Models to Study Mitochondrial Function in Neurological Disorder“, Annesley and Fisher explained why they’ve turned to cell lines to study mitochondrial functioning. In the review, they explain that while system-wide mitochondrial dysfunction would be most like to show up first in tissues with higher energetic needs like the muscles and the neurons, it should still be evident in all types of cells – including cells in the blood that are easier to get and work with.
Fisher believes the cells he’s using – continuously proliferating lymphoblastoid cell lines (LCLs) to which Epstein–Barr virus (EBV) DNA has been added – are perfect for assessing mitochondria. When used in his matter, EBV will make very small changes to the cell but will prompt them to become activated. It’s the ability to analyze the activated cell – and the burst of mitochondrial activity that comes with it – that Fisher was apparently after.
In the review article, Annesley and Fisher showed how LCLs have been valuable in understanding the mitochondrial issues in neurological diseases like Parkinson’s Disease, Huntington’s Disease, Amyotrophic Lateral Sclerosis (ALS), Fragile X-Associated Tremor Ataxia Syndrome (FXTAS), and ME/CFS.
They noted that the direct measures of mitochondrial energy production done in non-proliferating, quiescent cells, such as PBMCs and muscle cells, have, thus far, revealed no clear, consistent differences in ME/CFS. It was the use of LCLs, Fisher believes, that enabled him to find a “clear and specific defect” in the mitochondria of ME/CFS patients.
“A Clear and Specific Defect”
Their first major study, “An Isolated Complex V Inefficiency and Dysregulated Mitochondrial Function in Immortalized Lymphocytes from ME/CFS Patients“, indicated that oxygen wasn’t being well utilized by the ME/CFS patients’ mitochondria; that relative to the amount of oxygen they were getting, the amount of ATP they were producing was low.
When enough protons are found in the intermembrane area (blue), Complex V should convert ADP to ATP. Fisher’s studies indicate it’s having trouble doing that in ME/CFS.
At the same time that was happening, several aspects of mitochondrial functioning were elevated. The first complex in the electron chain (Complex I) was working overtime, maximum oxygen consumption and maximum nonmitochondrial oxygen consumption rate were increased. Plus, the upregulation of the mTORC1 stress signaling complex associated with Complex I, the overactive fatty acid transporters, and the enzymes that break down that fuel so the mitochondrial can use it and power the Krebs cycle, suggested that the ME/CFS patients’ mitochondria were working awfully, awfully, hard.
The authors proposed that all that work was an attempt to compensate for damage to the last Complex (Complex V) in the electron transport chain where ADP gets turned into ATP. The compensation worked – but only up to a point. The ME/CFS patients’ cells were probably doing fairly well at rest, but once stressed – they were running out of gas.
Altered Diet for ME/CFS Patients’ Mitochondria?
That hypothesis in hand, they tested it. In their 2021 paper, “Dysregulated Provision of Oxidisable Substrates to the Mitochondria in ME/CFS Lymphoblasts“, Missailidis and Fisher looked to see if the mitochondria were getting the fuel they need to thrive. Once again, they compared lymphoblasts from people with ME/CFS (n=34) to healthy controls (n=31).
Glycolysis Hypothesis Down – Fatty Acid and Mitochondrial Dysregulation Hypotheses Up
“The potential for abnormal utilization of fatty acid β-oxidation has arisen clearly in our own work.” Missailidis et al.
Normal levels of glycolytic enzymes, plus former findings suggesting that the normal glycolytic rates and reserves are present, suggest that the glycolytic pathways are operating normally; i.e. glucose was being metabolized correctly and providing normal amounts of “food” to the Krebs or TCA cycle.
Instead of the cells turning to other sources of energy because glycolysis – the glucose pathway – is damaged, Fisher now believes they’re doing that for two reasons: in order to compensate for damage to the Complex V in the electron transport chain, and in response to increased levels of cellular stress; i.e. a “dysregulated energy stress signaling” process.
This study’s results concurred with those of past studies which suggested that fuels other than glucose – which provides an easy and clean source of energy – such as fatty acids and amino acids, are indeed being preferentially used to produce energy in ME/CFS patients’ cells.
Activated Fatty Acid Oxidation Pathway Stands Out
One energy source, in particular, stood out – fatty acids. B-oxidation refers to the process by which enzymes break down long-chain fatty acids in the mitochondria to get them ready to be used. for fuel; It was remarkable to see that almost half (9/20) of the most significantly upregulated pathways “pertained directly to fatty acid β-oxidation”.
Overall, the mitochondrial issues were highlighted to a remarkable degree. Besides the innate/adaptive immune regulation, the significantly upregulated pathways all involved the mitochondria. They included the TCA cycle, respiratory electron transport, other mitochondrial proteins, and the pentose-phosphate pathway.
Enzymes involved in peroxisomal B-oxidation were also upregulated in ME/CFS. The peroxisomal fatty acid-b-oxidation finding was intriguing given Ian Lipkin’s latest study, which suggested that peroxisomal dysregulation plays a key role in the mitochondrial problems in ME/CFS.
Interestingly, the mRNA in the ME/CFS patients’ cells did appear to be telling the glycolytic enzymes to pick up the pace a bit, but the protein analysis indicated they didn’t do that. This is what happens, though, when cells begin favoring fatty-acid B-oxidation instead of glycolysis.
All of this strongly “suggested that an upregulation of fatty acid β-oxidation was occurring in ME/CFS patients’ cells.”
Still, there were questions. While this study overwhelmingly supported the idea that fatty acids are increasingly being used for fuel in ME/CFS, some study results suggest that fatty acid oxidation is actually being hindered. One study which specifically looked for increased fatty acid utilization in muscle cells did not find it. The authors had a possible answer for that but asserted that more direct measures of fatty acid β-oxidation utilization in lymphoblasts, as well as protein expression in muscle cells, need to be done.
Branched-Chain-Amino-Acids (BCAAs) Getting Chewed up
The mitochondria’s need for more and more fuel (electrons) appears to be causing them to break down the BCAAs as well. As the mTORC1 complex stimulates BCAA breakdown, and the mTORC1 pathway was chronically activated, this was expected. The authors also asserted there was “compelling evidence” that proteolysis – the breakdown of proteins – in order to release amino acids for fuel was occurring in ME/CFS.
Complex V Problems Are Key?
“More importantly, our observations here support our previous proposal that upregulated fatty acid β-oxidation in ME/CFS cells provides acetyl CoA to the TCA cycle more rapidly, provisioning the upregulated respiratory complexes with reducing equivalents to accelerate respiration and compensate for inefficient ATP synthesis by Complex V.” The authors
.jpg)
Rapamycin (apparently in red) inhibiting MTor pathway. (From Enzymylogic via Wikimedia Commons).
Why, though, were ME/CFS patients’ cells choosing to rely on fatty acids or amino acids for fuel when glycolysis provided such an easy pathway to energy?
If I have it right, the authors believe this is all an attempt to make up for problems in the final and determinate step of the electron transport chain in Complex V, where ADP is converted to ATP. The reason fatty acid metabolism is being emphasized is that it operates more quickly than the other pathways – and Complex V is screaming for more fuel.
Far-Reaching Consequences for Glutamine-mTORCI-Rapamycin Connection?
Another possibly critical finding concerned the elevated activation of enzymes involved in breaking down mitochondrial glutamine. This finding could explain the reduced levels of glutamine found in several studies and suggested that the mitochondria in ME/CFS patients’ cells were also preferentially breaking down the glutamine amino acid for fuel. The authors believed that this metabolic dysregulation could have “far-reaching consequences given its importance in many cellular processes”.
Glutamine degradation activates mTORC1 (mechanistic target of rapamycin complex 1) signaling and, as noted, chronic mTORCI activation was also found. The mTOR complex is a nutrient/energy/redox sensor and controller of protein synthesis.
The Gist
- It took Paul Fisher of LaTrobe University in Australia ten years to get to ME/CFS, but when he did he made the most of it.
- Using a novel technique using immortalized immune cells, Fisher and his lead researcher, Daniel Missailidis, have unveiled a series of significant abnormalities in the mitochondria of people with ME/CFS.
- Problems with the penultimate complex in the electron transport chain (Complex V) appear to be causing ATP production to decline dramatically when the mitochondria are put under stress.
- Despite a series of compensatory reactions, the ATP production of the mitochondria appears to decline by 25%.
- While problems with glycolysis – long thought to be present in ME/CFS – have not shown up – glucose is being shunted to a different pathway than usual. More importantly, Fisher found that the mitochondria in ME/CFS are preferentially turning – perhaps in an attempt to feed a broken Complex V – to amino acids, and particularly fatty acids.
- The authors pointed to evidence proteins were being broken down for fuel in ME/CFS. Glutamine degradation, in particular, was highlighted. Glutamine degradation triggers the mTORC1 pathway.
- Inhibiting the mTORC1 pathway has been associated with longevity in several animals. The authors did not recommend Rapamycin (or any other treatment option), but it has been used to inhibit the mTORC1 pathway.
- “Compelling evidence” also suggests that higher than normal numbers of cells are being broken down as well.
- The authors cited numerous studies they believe should be done to further understand the mitochondrial problems found in ME/CFS.
The search has been on to find ways to tamp down mTORC1 activity. Some dietary compounds (resveratrol, curcumin, caffeine, and alcohol) appear to be able to, and rapamycin (Soriolimus, Rapamune) – a drug that has elicited some interest in ME/CFS – has as well.
The authors asserted more work on mTORC1 activation in ME/CFS should be done. Plus, the amount of glutamine possibly being degraded indicated that studies focused on glutamine metabolism are needed as well.
The authors also asserted “compelling evidence” that the ubiquitin-proteasome system that attacks and breaks down cells is upregulated in ME/CFS patients’ cells as well.
Conclusions
Fisher has put together a set of novel studies that suggest that problems with glycolysis are not to blame but that something in the last complex of the electron transport chain has gone haywire in people with ME/CFS. (Fisher did find that glucose was being shunted through the pentose-phosphate pathway, though).
Fisher’s studies are different in that instead of assessing serum or plasma, they’re using immortalized lymphoblasts in the laboratory. This means the mitochondria are not exposed to factors in the blood which could be impacting them. On the other hand, this method appears to be providing a purer test of mitochondrial functioning and has been used to good effect in neurological diseases.
Fisher is the first that I know of to identify a deficiency in a specific complex in the electron transport chain. His prior study found a 25% drop in ATP production by Complex V when it was put under stress. That drop apparently triggered a wide range of compensatory reactions: extra copies of mitochondrial complexes were created, Complex 1 activity skyrocketed, proton pump activity increased, as did enzymes that consumed oxygen. All that compensatory activity was able to maintain the mitochondrial production at rest – but not when the mitochondria were put under stress.
This study is the last of several which have suggested that the fuel utilization by the mitochondria has shifted dramatically in ME/CFS. Instead of glucose, fatty acids, in particular, but also amino acids (BCAAs), are being preferentially broken down to provide fuel.
Fisher believes ME/CFS patients’ cells may be seeking out other fuels in order to compensate for the striking deficiency he found in Complex V. A chronic elevation of the mTORC1, and perhaps AMPK, may be adding to the mess. Except for the possible use of Rapamycin or other mTORC1 drugs or food sources, the consequences of this study for treatments are unclear. (The authors made no treatment recommendations and noted that more study is needed.)

Fisher’s new technique has unveiled some intriguing anomalies in mitochondrial energy production in ME/CFS.
It’s not clear, at least to me, how the problems utilizing long-chain fatty acids that have been found in some fibromyalgia patients relate to this study. This study’s findings, on the other hand, seem to fold in well with those from Ian Lipkin’s latest study, which suggested the peroxisomes may play a key role in the energy production problems in ME/CFS. Fisher’s study suggests, but does not prove, that the mitochondrial problems found in ME/CFS may be present system-wide; i.e. they could be present in every cell of the body.
With Fisher, we got a new approach to studying the mitochondria – which thus far has unveiled new and dramatic possible abnormalities – and may be clearing up some issues in the mitochondrial field as well. Fisher’s studies have also opened up many new opportunities for study as he and Missailidis, again and again, pointed out areas that need investigation.
- Coming up – A Mitochondrially Based Diagnostic Tool for ME/CFS?



I’m a participant in Paul Fisher’s mitochondrial studies. I’m hopeful that one day the 3 pathology tests he discusses in another paper may actually come to pass – provided they’re adequately funded.
BTW, you describe Daniel Missailidis as Paul’s head honcho. Paul runs the lab, Sarah Annesley is a research fellow and Daniel is their post-doc researcher. I’m sure Daniel would appreciate the promotion (!) but Paul may not ☺
Thanks for the insight. It’s always hard to know who to acknowledge. Fisher is the one behind it all but I always want to acknowledge the lead researcher in the study as well – which has been Missailidis in all these studies. He’s also presented the data – quite impressively – in conference presentations. (I changed “head honcho” to lead researcher.)
Fisher and Annesley produced the lymphoblast overview.
Fiona, is there a link to the paper you refer to re: three pathology tests?
are the tests related to activin or creatinine clearance ( urinary) such as in this 2019 paper?
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6787626/#__ffn_sectitle
“Rethinking ME/CFS Diagnostic Reference Intervals via Machine Learning, and the Utility of Activin B for Defining Symptom Severity
Brett A. Lidbury, Badia Kita, […], and Mark Hedger”
I am also aparticipant in this study. This study has been going g over three years and we in Victoria Australia are willing to help our researchers in any way we can.
So grateful to Daniel and Sarah for visiting and allowing us to help from home
+I am involved in using PBM to actively treat CFS?ME? Fibromyalgia and Flus…., I have a volunteer in Melbourne who had all the above diseases from 1992 till 2019. He is willing to volunteer for any Medical Tests regarding his prior condition and to the changes that has occured. . He is well now and can indulge in strenuous exercises, has his life back and does not suffer from PEM. I would like to invite a serious investigation into an actual subject. It appears that there had been changes in the composition of his gut microbiota. He is now able to eat well and convert his food into ATP.
Hi. I am doing a study on interconnected diseases at UMAsS Chan on US. It is called Reclaim and on my website. Could we connect?
What does PBM stand for?
Hi Cort, thank you for featuring the work of Paul Fisher here in Australia.
I’ve sent you a small donation in honour of his work.
Cheers,
Gail
Thanks Gail – I certainly can’t assess the value of one technique over another but FIsher has clearly been plowing a lot of new ground. 🙂
I wonder how this and Ian Lipkin’s findings might fit in with OMF findings that the plasma of an ME patient can convey the disability to another subject — and, conversely, that plasma from a healthy subject “perks up” function in a diseased subject. (I’m not clear if participants in these trials are actual humans or simply cell lines –it’s been a long time since I saw Ron Davis present this information.)
Ron Davis, Fluge and Mella and Prusty have all found indications that something in the blood is having an impact. They’re not to my knowledge using human subjects but Davis and I think Prusty has as well found that things change in the lab when plasma/blood from ME/CFS patients is introduced. Hopefully, at some point someone will find that substance.
Some think the mitochondria might be damaged by that substance and that damage would show up when the mitochondria are isolated.
Do you have a link? I want to show this to my docs and my wife’s docs (we both have me)
I’m not sure if this is relevant, but when I looked up ‘ Lymphoblastoid’ I saw they were also used to study and find Mitochondrial Dysfunction in Autism. Which I remember Bob Naviaux was Also studying (both autism and ME/CFS)
I suspect that Autism is ME/CFS but only when in the prenatal and early years of essential neuronal brain development
Naviaux’s ‘Cell Defence Response’ hypothesis was that the immune system was putting cells into a hibernation state to preserve energy when the body thought it was under attack by infection or injury. It makes sense as a mammal can’t go out and gather the main fuel needed i.e. carbohydrates when sick or injured, so the cells convert to using stored energy, i.e. fats and amino acids from the breakdown of muscle proteins. This state however seems to lock in for the rest of the lives of people with ME/CFS (I suspect an ongoing autoimmune attack).
In Autism however I’m wondering if because the child is so young that probably the autoimmune issue eventually resolves reasonably early (as we often see older children with ME/CFS), so their energy returns, but because they missed that first 3 years of essential neural pathway development, that their brains never fully develop. You will often find older kids with autism have plenty of energy. Although too much brain stimuli, (audial and visual), will tire them out quickly.
I know a few autistic children all older than 3 years, who don’t get PEM either. And a 9 year old has improved dramatically. Although still has difficulty with ‘Theory of mind’ (understanding others) his mother said his energy had increased a lot early in his infancy (before that crucial 3 year mark) he is now high functioning.
It’s like the children with autism, have somehow recovered from energy issues of ME/CFS but been left with underdeveloped brain function.
Or have people with Autism been more easily able to utilise the fats and amino acids for fuel because it happened to them so young in life? I presume some people with autism may stay locked in a low energy cycle too, although I haven’t heard of it.
Does anyone knows if this overlap between ME/CFS and Autism is still being looked at?
Is it confirmed if their metabolism returns to normal as they age? If so, what caused that reversal back to normal glucose energy use, could help ME/CFS research.
Further, Myself as an adult with ME/CFS from the age of 17 I found maturing socially suddenly very difficult.
Brain Fog and suffering from too much sensory input as an adolescent or adult with a developed brain is hard enough, but imagine having all that as a baby, when one is meant to be learning the most important cues and keys to life via person to person interactions.
No wonder autistic children regress into themselves.
Many adults with fully developed brains and severe ME/CFS will know exactly how it feels to escape from the noisey chaotic world. I think these two unexplained conditions are actually infant and adult versions of the same disease.
Brendan, i for one think you are hitting a possible linking feature of autism and cfs/me.
Thank you for pitting your thoughts ‘out there’
cheers
Brendan I agree with you 100%. The BCAAs are interesting. My son was autistic. As part of his healing I discovered that animal proteins of any kind destroyed his speech when he was a toddler. So a nutritionist gave him BCAA that I fed to him several times a day to replace all animal proteins. He recovered over the course of the next two years. He is a normal happy teenager now. I believe we were only successful bc we switched to no animal proteins during his early toddler to age 5 years. Had we waited or never done this, he would be seriously impaired today. His body needed BCAAs but could not properly utilize the animal form of proteins. I do think there is similarity with mecfs, whether it is fatty acid, protein, or glycolysis. We may need the simplest forms of these to feel better.
I have immune dysregulation, a tendency toward chronic infections, immunodeficiency and autoimmunity. And hormone and nutrient deficiencies, and mito dysfunction.
But, none of my symptoms are anything like autism. Nor are those of many other ME/CFS patients
I posted before seeing this. Do u believe there is muscle loss due to using them for fuel? I have lots of muscle loss and not sure it is due to de-conditioning.
My family has a history of both Autism and ME/CFS. I believe there maybe a familial link and would be interested in any follow up on both topics
Ah, Cort you’ve done the work for me!
To explore why Fisher/Missailidis & others obtained different glycolysis results I’ve been looking for a (free) copy of Armstrong’s 2015 paper.
Another rabbit hole conquered.
Thanks!
🙂
I wonder what this would mean for treatments? My endocrinologist has put me on a concentrated activated fish oil ‘SPM Metagenics’ and ‘MitoQ’ which I purchase from New Zealand. It’s taken a few months to feel a difference, but I’m feeling a little more power in my muscles and the weight I gained over the last four years has dropped back to the weight I was for most of my adult life. I still crash with PEM, however, I now get a couple of hours per day where my muscles feel normal if I don’t overdo things. I forgot what that felt like to be able to walk from room to room without feeling like I’m wading through wet concrete. I’ve also been taking a couple of other supplements to support mitochondria and metabolism, but I’ve been on them for years and they haven’t been nearly as effective.
Amanda, really interested in your regime if you are interested in sharing amounts of supplements taken.( no worries if not)
MitoQ has some really interesting attributes on renal tissue ( where they say is high mitochondria levels) according to this article:
“The targeted anti‐oxidant MitoQ causes mitochondrial swelling and depolarization in kidney tissue
Esther M. Gottwald, Michael Duss, […], and Andrew M. Hall”
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5880956/
hoping their findings lead to further discoveries of what the swelling is doing, and if it is beneficial for cfs/me patients.
Hi I’ve got kidney disease as well as ME/CFS/Fibro and a wealth of other problems.
I take mitra q should i stop if it is harming renal tissue.
For those that want to avoid the fish part of fish oil, i.e. environmentalists vegans, people wanting to avoid contaminants, or those wanting to avoid fishy burps lol, or those wanting to not support the now known widespread slave labour in the fishing industry, or cruel fishing methods etc there is a clean safe alternative… algae oil…
Marine Derived Algae Oil is what the fish eat in the first place to get these fats. A good way to cut out the middleman and go straight to the source for Omega 3s
Algae oil also contains both EPA and DHA
everything comes together with unflexible Erythrocytes plus latent ACiD bloodcirculation
thats the cause of 265 symptoms
we urgently NEED those Lab Tests
Somaya, I couldn’t agree more!
https://www.latrobe.edu.au/news/articles/2019/release/chronic-fatigue-syndrome-research-funded
Above is an earlier article about this research and how a particular funding source made it possible—an interesting read.
I feel excited about this line of research!
“Here at La Trobe we have discovered a specific defect in the mitochondria – the ‘powerhouse’- of the cells of ME/CFS patients and are working towards a simple diagnostic blood test,” Professor Fisher said.
A blog on this is coming up. )
Cort,
I live in Melbourne. I am interested in contacting this group to be a participant, do you have their contact details?
A simple diagnostic test would be well appreciated.
All very interesting.
Fortuitous that the funding ramped up early in the covid pandemic before anyone had heard of ‘long covid’!
I say a this tongue in cheek… of course all of you knew about long covid… many of you are post virally challenged… so you all know about long covid. Even before you knew about covid!!!!
Yes, indeed. I hope everyone with long COVID understands that they’re actually joining a community of people with similar if not identical illnesses who’ve been fighting for recognition and funding for decades.
I certainly understand this! I’d be in much worse shape now if I hadn’t listened to the wise counsel of those who’ve Long struggled with ME/CFS. Everything I know about the importance of rest, pacing, and staying within my energy envelope comes from the ME community.
I was looking at complex V in the brenda enzyme database. Complex v dysfunction is implicated in a lot of systemic diseases and viruses and bacterial diseases. https://www.brenda-enzymes.org/enzyme.php?ecno=7.1.2.2&onlyTable=Disease There are a few activating compounds identified. https://www.brenda-enzymes.org/enzyme.php?ecno=7.1.2.2#SUBSTRATE If they really figure this out, they are likely to have a big impact on many diseases.
From what I understand, the mitochondrial complexes are constantly being remade. So if they are broken, is this process broken? Or is a signaling chain shutting them off or damaging them? Or is some other process damaging them? The post exertional malaise reaction is so consistent in how it starts and slows down and stops. And the timing is so weird – 24 hours to full on, 5 days or more to full off. I’m leaning towards a signaling reaction because of the timing strangeness and consistency. Wish I could do more than just guess.
A nice start, Chris 🙂
Interesting! For me PEM is exactly 5 hours post exercise. I can play sports no problem but 5 hours after wham brain fog fatigue sunken eyes confusion mood drops etc. Last for exactly 24 hours from onset. So strange! What happens 5hrs after exercise , wish I could monitor somehow
When I think about mitochondria, I wonder whether an adaptation could be created in humans, the way that something occurred in a sea creature.
The creature in this particular case is Elysia timid — and there is an article here: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7679131/
Elysia is a sea slug . . . but it’s a small, charming thing that looks a lot like a leaf floating on or in the water.
Here is one picture, but of course there are others:
http://www.sci-news.com/biology/science-eastern-emerald-elysia-genes-algae-plant-02466.html
And it has somehow become able to ingest plant life, and “steal” chloroplasts. And managed to have those continue to work — for a while — inside itself, Elysia.
If humans could steal mitochondria . . . or even chloroplasts . . . and have them continue to function for a while.
I took Rapamune for 1 month, did absolutly nothing. Another lame duck piece of research sadly.
My testing shows:
– impaired fatty acid oxidation
– preferential use of glycolysis
– overworked complex IV
– high glutaric acid
– high normal free any total carnitine
I get burning/drained feeling in legs and forearms after 2 minutes of increased activity or aerobic exercise, even though I can walk a few miles slowly without inducing PEM.
I’m skeptical of the choice of cells Fisher’s team are using to decide these things. I’ve been to 4 mito specialists and though they agree something is wrong, they are clueless, as I don’t have a primary (genetic) mito disease.
BCAAs and glutathione help me about or reverse PEM consistently. My doctor has had me try oxaloacetate and sirolimus. I can’t say if they do much or not.
I also take nicotinamide mononucleotide (NMN) and riboflavin-5-phosphate (to support FAD).
Would definitely like to see more research in this area.
Interesting – preferential use of glycolysis. If I understand it right low glucose levels in the metabolomic studies suggested preferential use of glycolysis but Fisher says that is a normal outcome of preferential fatty acid oxidation. Your problems with fatty acid utilization seem similar to those found in the recent fibromyalgia blog.
I guess time will tell with Fisher’s method. Some have suggested that the glycolysis issue may be caused by something in the plasma.
Interesting, I can exercised no problem but PEM kicks in exactly 5 hours after for 24 hours . I can’t stop it with any supplement so far. How did you test the above learner? Thanks
Deep breathing & deep sleep will take the edge off that 5 hour mark or sooner post exertion . It is a hallmark of fibro .
Slow down don’t push the envelope . Stretch a lot to release buildup in muscle fibers. Acupuncture is my go to 🙂
Your description ‘I get burning/drained feeling in legs and forearms after 2 minutes of increased activity or aerobic exercise’ really hit home. The leg burning is not something that seems to be common. With me, stairs often cause this burning in the muscle, and I need to rest to allow it to subside a bit, but once I resume, the pain comes back rapidly. I’ve described it as muscle burn, oxygen deprivation since it occurs on exertion. It seemed oxygen related, in my mind at least, so much so I convinced my primary care to approve me for the icpet testing here in Boston with David Systrom thinking it would prove conclusive.
It did not. https://www.healthrising.org/blog/2019/12/11/oxygen-extraction-post-exertional-malaise-chronic-fatigue-syndrome/
Maybe the metabolic anomaly is part of the sickness behavior? People sick with flu have similar problem generating energy after all. It would’ve been more interesting if they compared with cold/flu patients rather than healthy controls. But I can see that would be difficult to arrange since cold/flu last only a week. And there is no Chronic Flu-like Sickness other than CFS that I can think of.
With ME/CFS, I do naturally gravitate towards a low carb diet, but also towards a low fat diet. I think one article which I cannot remember said no direct relation could be found to dietary behaviour though.
5 years ago (at about moderate ME/CFS) I had ATP blood tests done and they were not particularly low (first test in the low part of “normal” range, second test from another lab 2 months later in the “normal” range).
I keep having muscle loss (at least in part due to de-conditioning). But I wonder if my body is breaking down muscle cells for fuel (amino acids). Is that how the body attains amino acids in such dysfunctional energy production?
I am interested in this as well… I have ME/CFS like symptoms as part of long haul Covid. I have been bedridden for over two years with severe fatigue, PEM, and neuro issues. I did notice in the early days of my illness, I rapidly lost a ton of muscle mass, way more than could have been attributed to just a reduction in activity levels. I have seen other people in my long haul groups talking about a similar issue.
It is due to catabolism. I have the same problem. You can’t train your muscels. Keep in the envelope don’t push to hard. Do what you can if possible to avoid de-conditioning.
I wonder if taking amino acids would help, so that is used instead of muscle.
I can exercise and play sports for 1-2 hours, feel good after. Exactly 5 hours later wham PEM brain fog heavy eyes swollen belly (maybe from cortisol?) fatigue etc. Lasts for 24 hours from onset every time. Strangest thing . Have no idea where to go from here
I wonder whether there are any supplemental ways of enhancing Complex V if that is indeed the stage of mitochondrial function that is breaking down? Aerobic exercise enhances mitochondrial enzyme levels but of course that is difficult for many.
as a person with 30 years of ME/CFS problems, I would find it extremely helpful if any of the above conclusions could be summarized in a way that my “limited brain” could get it..
Is there something, anything I can do to get better?!
The first 40 years of my life I was in very good health and very fit.
In 1992 we bought an old run down house on 10 acres. The house was greatly infested with flies and fleas and we were advised to have it sprayed by professionals.
At the same time in order to clear the land for a large vegetable garden and to rid the pasture of thistles and obnoxious weeds Glyphosate (RoundUp) was used too.
Within three months I gravely very ill. It felt like an awful, awful virus attack with all the symptoms of a nasty Flu, high fever, very, very weak, aches and pains all over, impaired cognitive thinking, forgetfulness, very bad cough, stiff painful muscles, awful backpain, terrible headaches, sore throat and extreme chemical sensitivity. For months and months no improvements, digestive and gut problems were now also problematic. I became bedridden.
The months turned into years with little change.
After years of despair, on the 12-1-2004, I decided to follow Hippokrates’ advice:
“Let your food be your medicine and your medicine be your food.”
With help of the family I started to make fresh vegetable juice and drank daily one liter accompanied by a diet of 80 raw vegetables and fruits, then also pulses, brown rice, lentils and nuts seeds as proteins. No white flour, no sugar except what was in the vegetables and fruits, no animal protein, no dairy, no coffee. For the first two years all of my food had to be blended and eaten with a teaspoon otherwise I couldn’t digest it. But, over time my physical and mental state of health improved more and more. After 15 years I was now 70 per cent better, except that ever since those first chemical poisoning during winter 92, I can no longer tolerate any herbicide spraying: in within 12 hours I get extremely fatigued, get headaches, muscle pain, back pain, nerve pain, sore throat, sleeping problems, brain fog etc.
I still had to be careful with physical activities but I was walking and up and going, able to do all the housework even shopping and outings, and even travels overseas were possible. My brain also was so much better. I was able to listen to conversations, lectures, music, watching TV, again all of this with care. Now life was good. I could get off anti-depressives, but still needed help with muscle relaxants before bedtime in order to go to sleep and more or less stay asleep.
Then came the temptation to go easy on the strict health diet.. Five years ago I started to add e.g. some white flour (pasta), some coffee (1 cup a day), a little bit of chocolate, biscuits, but the worst mistake: stopped juicing.. I was still “fine” but not as well as before when I strictly followed the successful but rigid diet.
Last October I had the first Covid vaccination (Pfizer) and in within 24 hours many of the ME/CFs were back: ongoing severe headaches, forgetfulness, brain fog, enormous fatigue, aching body: inflamed stiff muscles, Fibromyalgia, Neuropathy, severe lower back pain, no tolerance of noises of any kind, the circadian clock messed up. The greatest problem is the fatigue. I haven’t been able to go for my daily walk since October. After four months was told it was high time to have the second shot, I am now really struggling.. The greatest problem is the fatigue. I now need at least two bed rests during the day. –
Perhaps all of this can also be helpful in a small way to shed some light on the complex problems with ME/CFS
With regard to quick conclusions – the Gist is about as good as it’s going to get. Most of the research cannot be translated into easy treatment recommendations. Thanks for providing your story with your diet though. 🙂
Cort,
I am going to hazard that the mitochondrial bottleneck is
Acetyl CoA.
Rob Phair talked about sourcing Carboxyl groups in the TCA cycle. Please recall that my initial theory of CFS muscle exhaustion was that Acetylcholine was either underproduced, or under-received by receptors. In the case of MG Myasthenia Gravis, the latter is the case. I think that is where Mestinon helped one Recovery.
But Acetylcholine reuptake, lysis and recreation depends on sufficient Aceytl CoA in the first place. That there requires sufficient Acetate production in the gut in the first place, plus sufficient absorption of Choline (B vitamin), also in the gut.
Thus dysbiosis would appear to be huge in the causation of the conditions for developing CFS . A poor microbiome will not create the Butryrate needed for the intestine;s own fuel needs, nor will it create enough Acetate for the muscles’ and cognitive brain cells’ need for Acetylcholine to trigger exercise.
I guess that if the muscle cell can no longer depend on Acetylcholine, then it can no longer exercise.
But what it CAN still do is to generate heat,
through irisin release.
In this way, the body maintains its core temperature, but does not allow exertion