

Geoff’s Narration
The GIST
The roles autoantibodies are playing in long COVID (and ME/CFS) is one of the great questions in these diseases. There’s never been an autoantibody study like the Akiko Iwasaki-led 40-plus author, “A causal link between autoantibodies and neurological symptoms in long COVID“.

Bingo! It’s now clear that autoimmune processes are alive and well and causing mischief in a large subset of long-COVID patients.
Note the word in the title – “causal”. “A causal link between autoantibodies and neurological symptoms in long COVID”. That’s a big change – and the editors of the Cell medical journal – one of the most prestigious medical journals in the world – gave it their OK. That’s a big deal! We’ve been talking about and questioning autoimmunity. Is it happening? Is it not?
Now this study says “yes, it is definitely happening”, and that will ultimately open up a wide array of treatments that haven’t been open to us before. We’re still not at the treatment stage; that will take identifying the precise autoantibodies at play (not an easy task), but it appears that the first big step has been taken.

As we’ll see, one of the findings has profound implications for autoimmunity in ME/CFS as well.
THE GIST
Antibodies affecting blood vessel functioning were found in long COVID – and helped to validate an ME/CFS subset as well 🙂
- The roles autoantibodies are playing in long COVID (and ME/CFS) is one of the great questions in these diseases. These immunoglobulins produce autoimmune conditions by mistakenly attacking our tissues.
- Infections often produce dramatic upsurges in autoantibody levels but they almost always die down quickly without causing problems. That didn’t happen in long COVID.
- Note how firm the title of the paper (“A causal link between autoantibodies and neurological symptoms in long COVID”) it is. It states it found a “causal link” between autoantibodies and neurological symptoms in long COVID. That’s something we haven’t really been able to say before and it’s a big deal coming from one of the top medical journals (Cell) in the world.
- The study found more autoantibodies and a more diverse set of autoantibodies which tended to target tissues from the central and peripheral (body) nervous systems on the long COVID patients.
- When they exposed the IgG antibodies from the long COVID patients to nervous system tissues what they found made perfect sense with what we know about long COVID and ME/CFS. These autoantibodies strongly interacted with/attacked parts of the brain associated with autonomic nervous system regulation, sleep, pain), the sensory system, and interestingly enough with cells that affect the blood vessels that wrap themselves around and protect the blood brain barrier.
- The authors were not able to show which specific autoantibodies caused which specific symptoms, but the association between overall antibody levels and symptom load in long COVID was clear.
- When they tried to determine if they could produce autoantibody subsets a subset focused on autoantibodies that affect the blood vessels and the microcirculation, in particular, and autonomic nervous system functioning popped out.
- This was a big win for the ME/CFS field as German researchers have been studying this same subset in ME/CFS for years.
- The mouse transfer studies which transferred long COVID antibodies (IgG) into mice produced eye-opening results. Simply given IgG to them essentially turned the mice into long COVID and/or ME/CFS mice; i.e. fatigued mice with increased sensitivity to pain, cognitive, sensory, balance and coordination problems.
- Once again, the areas of the brain the IgG antibodies attacked were reminiscent of those seen in long COVID and ME/CFS imaging studies. These areas of the brain are designed to keep you fatigued, hypervigilant, to make you perceive stimuli as threatening, to lock in those memories, to be bothered by odors or sounds, to make movement effortful, etc.
- In other words, they’re designed to make sure that you’re as immobile as possible.
- David Putrino, one of the leaders of the study, reported that the study opens the door to “a number of effective treatments for autoimmunity that could significantly improve the symptoms of millions of people with this chronic condition.”
- We’re getting there but we’re not there yet. First, researchers are going to have identify which autoantibodies/sets of autoantibodies are triggering which symptoms in long COVID. (That is doable but not an easy task in diseases as complex as long COVID and ME/CFS. Once that happens they have a bevy of options to clear the autoantibodies out, to neutralize them, to effect their functioning, etc.
- It also appears clear that autoantibodies aren’t the only story in either long COVD or ME/CFS. While they appear to make a major difference in some patients, pathogen persistence, genetics, viral persistence, innate immune activation, latent virus reactivation, dysautonomia, and microclots will all ultimately play roles.
- The nice thing about the autoantibodies is that they exist in a well-developed field. We know how to find them and target them. Even if they are not the be all and end all, the right treatments hitting the right subsets could bring major relief.
Health Rising’s Donation Drive Update!

Is autoimmunity a question no more? For at least a large subset of long COVID patients, these researchers believe that question has been settled.
Thanks to everyone who has brought us over a third of the way to our goal.
This was a fascinating and encouraging blog because it addressed a central question in long COVID (and ME/CFS, for that matter) that Health Rising has been following for as long as I can remember: is autoimmunity a real thing? The successful outcome of this study made it a delight to report that it looks like it is.
Your support has allowed HR to track this subject over the years. So thank you! And please support us in a manner that works for you. There’s much more to come…
Health Rising is not a 501 c (3) non-profit
Study Results
“Collectively, our data illustrate the pivotal role of AABs (autoantibodies) as a key driver of neurological disorders in a subset of LC patients.” The authors
This study went out of its way to make sure it correctly identified its autoantibodies. For one, it used four approaches (immunofluorescence (IF), ELISA for GPCRs/ionotropic receptors, a >21,000-protein HuProt microarray, and mass spectrometry via antibody pull-down) to characterize and confirm the autoantibodies.
In both groups, antibodies sprouted like dandelions after a rain, but the long‑COVID patients displayed a more numerous and more diverse set of autoantibodies that were reacting with tissues, particularly central and peripheral nervous system tissues.
Infections typically cause a flare in autoantibody production, which soon dies down. The fact that these autoantibodies were still present over a year later in many of these patients strongly suggests that an autoimmune process was ongoing.
The autoantibody production across long COVID was quite heterogeneous, and this was expected in a disease that appears driven by numerous factors, including persistent virus, herpesvirus reactivation, tissue damage, and autoimmunity.
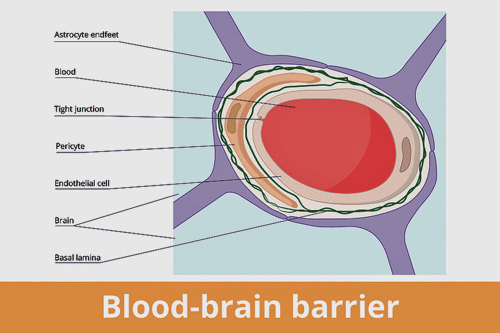
Autoantibodies appear to be attack the pericytes (orange section) surrounding the blood vessels that make up the blood-brain barrier – providing yet another possibility for interrupting blood flows – and allowing unwanted factors in the brain.
When they exposed the IgG antibodies from the long-COVID patients to nervous system tissues, they found that it attacked parts of the brain (locus coeruleus (brainstem – principle source of norepinephine, autonomic nervous system regulator, sleep, pain), thalamus (major regulator of sensory information (!), brain fog), meninges (pericytes/endothelium) (poor blood flows associated with reduced waste clearance, blood brain barrier problems, brain fog, headaches, neck stiffness), as well as endocrine tissues (low cortisol?) and a wide array of nervous system proteins.
The IgG from the healthy controls and recovered COVID-19 patients bound to these tissues to some extent, as expected. The IgG from the long-COVID patients reacted/bound to/attacked – take your pick – the nervous system tissues much more strongly.
The meninges/pericyte/endothelium finding was particularly interesting because it suggested that antibodies were binding to the pericytes and endothelial cells that regulate capillary tone, barrier function, and microcirculatory flow.
Microvascular Pericyte Diseases?
Pericytes are small cells wrapped around capillaries and other small blood vessels that stabilize them, control blood flows, and maintain the blood-brain barrier. They also help direct blood to areas of the brain that need more blood (neurovascular coupling) and even play a role in inflammation. Antibodies binding to pericytes could cause immune cells to attack them, producing inflammation, and problems with the microcirculation, in particular. A 2022 paper proposed that long COVID and similar diseases are essentially systemic (body-wide) microvascular pericyte disease.
Moderate Symptom Correlations
Being able to correlate autoantibodies with symptoms would be a big deal. If I remember correctly, Avindra Nath has pointed out that all infections cause many autoantibodies, which are often secondary manifestations of a disturbed immune system and nothing more. Showing that antibodies from LC patients correlate with symptoms would be a big deal. They got about halfway there.
They wanted to find if IgG from patients with a specific symptom was more likely to react with a tissue that caused that symptom; i.e., did a long-COVID patient with headache also have IgG antibodies that the former test showed were reacting with meningeal tissues.
They had some promising leads, but a statistical check knocked out most of the correlations. Part of the reason may simply be the complexity of long COVID. They found, for instance, many autoantibodies were associated with headache in long COVID.
When they looked at the global picture, though, they found consistently higher signals of autoantibody reactivity in LC patients and higher symptom levels in LC patients whose antibodies reacted against tissues.
Now that they’re clear that autoantibodies are causing symptoms, the next step is to identify the specific autoantibodies that are targeting specific tissues and causing symptoms associated with them.
An Autoimmune Autonomic Nervous System/Blood Vessel Subset in Long COVD (and ME/CFS?) Pops Out
Next, they wanted to see if they could identify subsets of LC patients who had distinct antibody profiles – and they found two of them. One was characterized by consistent positivity for beta-1 and beta-2 adrenergic receptors, endothelin A receptor, and muscarinic acetylcholine receptor M4.
Antibodies affecting blood vessel functioning were found in long COVID – and helped to validate an ME/CFS subset as well. 🙂
This was a fantastic finding for the ME/CFS field, in particular, because not only does it replicate similar findings in ME/CFS (and long COVID), but it also did so with highly regarded researchers in a top medical journal.
These receptors regulate autonomic nervous system function, blood vessel function, the microcirculation, and orthostatic intolerance, and have attracted considerable interest in ME/CFS because they could affect key issues in the disorder.
This finding indicates that, when it comes to autoantibodies, a subset of long-COVID patients look exactly like a subset of ME/CFS patients, and, importantly, it provides a mechanistic hook which indicates that these autoantibodies are causing symptoms.
This “GPCR‑autoimmune dysautonomia” subset appears to occur in about 25-30% of ME/CFS patients. (The LC paper does not say how big the subset was in the study.) Carmen Scheibenbogen and other researchers who have been digging into this subset in ME/CFS for years must have smiled when they saw this pop out.
The Mice Validation
We’ve talked a lot about IgG transfer studies in ME/CFS, FM, and long COVID, but this transfer study was something else. It’s the most comprehensive mouse transfer study done in these diseases thus far.
Transferring purified IgG from long-COVID patients into mice triggered fatigue‑like behavior, balance and coordination problems, increased sensitivity to pain, and, importantly, because no one has been able to determine why it’s occurring, small nerve fiber damage. Interestingly, IgG from LC patients with chronic pain increased pain hypersensitivity in mice (!).
The increased plasma neurofilament light chain (NfL) levels suggested that the autoantibodies are causing nerve damage. It’s an intriguing finding, in part because the RECOVER Initiative is assessing neurofilament light chain levels in its long-COVID patients.
The brain activity of the mice given LC IgG painted a pretty good picture of what’s been found in long COVID/ME/CFS/FM. Note that they didn’t stress these mice at all. We got a picture of what long-COVID IgG does to the brains of resting mice, and it wasn’t pretty.
Areas of the brain involved in touch and pain
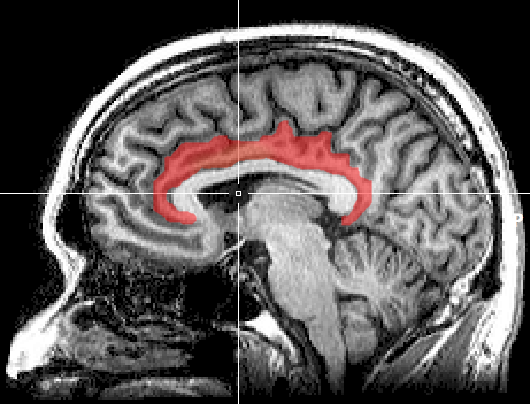
Antibodies attacked the cingulate cortex and other brain structures that have been identified in both long COVID and ME/CFS.
(somatosensory areas), that assess sensations in the gut and organ (visceral area), that carry sensory signals from the brainstem (medial lemniscus), and that track internal body sensations (insula) were lit up like a Christmas tree. So were other parts of the brain (primary auditory and visual cortices and associated areas) that determine how we respond to light, odors and complex visual environments.
If your entire body is in pain, if it seems like you can’t escape your body sensations, if it feels like your heart is pounding, if light, odors, and/or touch bother you … well, these are reasons why. The sensory pathways in your brain are overreacting to everything. It’s no wonder that Daniel Clauw calls fibromyalgia a sensory disorder.
The motor/effort areas of the brain were also lit up. Despite the fact that the mice were not stressed, were not moving, etc., the parts of the brain involved in planning and initiating movement (primary motor cortex), in controlling posture (reticular nucleus in the brainstem), and in integrating arousal and movement (taking action and moving) were stressed and activated.
This suggests that the “motor system” – the part of the brain that gets us to move – is confused, and is over-activated even at rest. It’s no wonder that mice given long-COVID IgG moved less – and no wonder that movement is often so effortful in long COVID (and ME/CFS).
We’re not nearly done yet, though. The long-COVID IgG also switched on areas of the brain guaranteed to make you feel bad and on edge. The anterior cingulate cortex (ACC) – which makes pain feel worse was lit up; the amygdala – the fear center of the brain – was on fire (nice!); and the claustrum – an integration hub – was in overdrive trying to integrate a bunch of apparently confusing signals. This triad of brain regions ensures that the sensory signals your brain receives are going to be perceived as threatening. This is where suffering comes to live.
The jacked-up hippocampal activity makes sure that you’re stuck in suffering mode: it’s what makes sure you remember what situations, smells, and activities provoked the pain and distress. If you did “X” and felt bad, it sticks that result deep into your brain and when that situation pops up again, like Pavlov’s dog, the alarm bells go off.
A hyperactivation of other regions (retrosplenial cortex, temporal association cortex, hippocampal CA1 and CA3) involved in attention, memory, focus, persistence, and location explains why people with long COVID often have so much trouble staying on track, get quickly fatigued when doing cognitive work, and can at times get lost even in their own neighborhood.
We still have one more area of the brain to check out. Hyperactivation of the midbrain (midbrain reticular nucleus, Edinger–Westphal nucleus, rostral linear nucleus of the raphe) disrupts arousal, wakefulness, sleep, and autonomic nervous system function.
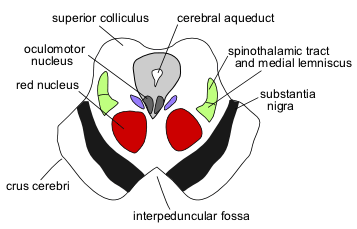
Disruptions to the midbrain could be impacting sleep, wakefulness, arousal, and the autonomic nervous system.
So, the brains of the poor mice given the LC IgG basically recapitulate what we see in long COVID (and ME/CFS/FM). The long COVID-IgG provided a recipe for fatigue, chronic pain, sensory problems, hypervigilance, cognitive problems, etc., even in unstressed, resting mice.
Throw in evidence of the microvascular blood vessel/blood-brain barrier problems, which could be driving all these issues, and you can see why the authors were confident they’d found at least some of the causal drivers of long COVID.
Given this study’s success and the passive transfer studies in fibromyalgia and ME/CFS, those two diseases will clearly be viewed in a more autoimmune light in the future.
Treatment Implications
“This new awareness of the physiology of long COVID will enable us to identify a number of effective treatments for autoimmunity that could significantly improve the symptoms of millions of people with this chronic condition.” David Putrino

The next steps – finding the exact autoantibodies/sets of autoantibodies at play.
This should bolster clinical trials aimed at reducing or neutralizing these specific autoantibodies in carefully defined subsets. The next key step is to develop a diagnostic panel that can sensitively and accurately detect the autoantibodies implicated in these diseases. The good news about ME/CFS is that a similar panel will likely be helpful.
“Before, we had no way of predicting who would benefit from therapies like IVIG or FcRn inhibitors,” he says. “Our study now shows that if you are in a subgroup of long COVID patients who have autoantibodies circulating in your body, this is a quantifiable sign that you may be a good candidate for these drugs.” David Putrino
Regarding treatments, the authors specifically focused on IVIG in the paper, noting that it’s clear that IVIG benefits subsets in long COVID and ME/CFS but that (again): a) there is an “urgent” need to develop biomarkers that can distinguish which patients will benefit from it; and that b) autoantibodies may provide those biomarkers.
Once those biomarkers are identified, a range of other therapies that can either degrade the specific autoantibodies (FcRn inhibitors), neutralize them (BC 007), temporarily remove them (immunoadsorption), or target antibody-secreting cells (e.g., anti-CD20 or anti-CD38 monoclonal antibodies), and CAR-T cell therapy, could be helpful.
The findings also highlight the need to find treatments that reduce blood vessel inflammation (complement inhibitors, pericyte/endothelial protectants) and improve endothelial and blood vessel functioning in a subset of patients.
In the end, then, no specific treatments are recommended, but the authors reported that a clear (if not easy) path to developing them exists.
Blood Donations Not Recommended
“In the U.K., having long COVID is an exclusion for donating blood, while in the United States, these individuals are still allowed to donate. Given the dangers that plasma from people with long COVID can pose for others, this country should be considering fundamental changes to its donation policies that reflect that health threat and are designed to fully protect the public.” David Putrino
The paper ended with a stunner. Because it’s now clear that some antibodies from long COVID are pathogenic, the authors asserted that the use of blood products from long COVID patients “requires careful consideration”, i.e., shouldn’t be done. One wonders how many people came down with ME/CFS and similar diseases after a blood transfusion.
Not the Entire Story
Autoantibodies are a piece of the puzzle, but even in the autoantibody subset, these authors do not believe they are the cause of everything long COVID. Would that it were so simple.

Autoantibodies will likely present part of the solution. They are compelling because they present a nice target.
While the evidence does suggest they are a major driver of symptoms in a major (or some major) subset(s), even in these subsets, factors such as genetics, viral persistence, innate immune activation, latent virus reactivation, dysautonomia, and microclots may also play a role. Damage to the blood vessels by microclots, for instance, could be driving the formation of pathogenic autoantibodies.
Antibody treatments may or may not get at the causal drivers of these illnesses, but they present a compelling possibility because we have good validation that they’re playing a major role, researchers know how to identify them and how to get at them, and the data thus far suggest that interrupting or removing them should help relieve symptoms.
That said, identifying them will not be an easy task in long COVID or ME/CFS. We have the tools, but as in the PrecisionLife study that Health Rising recently profiled, both long COVID and ME/CFS appear to be unusually heterogeneous diseases; i.e., we’re probably not looking at one autoantibody but combinations of autoantibodies. It will probably take large, well-defined cohort studies to pluck out the autoantibodies in play, and that will require ample funding.
A gold-standard study – which we probably won’t get – but which gets to the bottom of this once and for all, and includes both ME/CFS and long COVID, might involve hundreds of patients participating in a multi-step, multi-year project that costs maybe $5-$10 million.
The UK just pumped in $6.5 million for the SequenceME whole-genome study, though, and it was recently reported that Germany, get this, will be spending half a billion Euros (@$500 million US dollars) over the next ten years to study post-infectious diseases, so it is possible. Things will probably progress more slowly, but it’s possible.
If that post-infectious money comes through, Germany, not the US, not the UK, is going to be the world leader in post-infectious diseases – and this is not a small disease block. A blog on the NIH situation in ME/CFS, POTS, and long COVID is coming up. As usual, the NIH is pretty stuck in the mud, but there is hope. 🙂
Limitations
This was an exciting and fundamentally important study, but there were limitations. It was relatively small (n=55) and enrolled long-COVID patients infected with the original, most severe COVID strain. Because it was focused on a neurological subset of long-COVID patients, it may not apply to people with fewer neurological symptoms. (But really, how big could that subset be?)
Pet Long-COVID Study Peeve – Since we don’t have criteria that identify the neurological subset of long COVID or even for long COVID itself, once again, the “just who are these patients?” problem shows up. I’m sure that, given how schooled these researchers are in long COVID and how careful they were in the study, they recruited a good group of long-COVID patients.
But look at this lax criteria. To be enrolled in Yale’s MY-LC program, patients had to have “multiple (how many?) persistent symptoms such as fatigue, shortness of breath, cognitive issues, or other long COVID‑related symptoms” for more than 3 months after the infection.
In order to be enrolled in this neurological study, participants had to have a “high neurological symptom burden”. We never, though, get a complete list of neurological symptoms, and we don’t know how many neurological symptoms it took to have a “high neurological symptom burden”.
We do know that 63.6% of the participants had five or more neurological symptoms (brain fog 80.0%, headache 65.4%, memory loss 64.4%, dizziness 58.2%, sleep disturbance 58.2%, confusion 54.5%). Later, we learn that weakness, disorientation, tinnitus, dysautonomia, chronic pain (including inflammatory or neuropathic pain), and pain qualities like pins and needles, burning pain, and electric‑shock pain were probably included.
This kind of laxness is par for the course in long COVID, but it’s really unsettling coming from the ME/CFS field, where so much effort went into determining precise symptom criteria. Every long-COVID study may have its own inclusion criteria. Six years later, is this really the best the long COVID field can do (???)?
Conclusion
This was clearly a major study. Medical Express may have gone a little far when it seemed to suggest that the study showed that autoimmunity is causing all the symptoms of long COVID.
“A Mount Sinai-led research team has demonstrated that autoimmunity, in which the body’s immune system attacks its own tissues, is responsible for the often-debilitating and confounding symptoms of long COVID in a subset of people.”
David Putrino’s statement was a bit more circumspect (aka “a major contributor”), “now we have validated that autoimmunity is a major contributor to the symptom burden”, and that is a big deal.
We’ll see what happens next!
Health Rising’s Donation Drive Update!
Is autoimmunity a question no more? For at least a large subset of long COVID patients, these researchers believe that question has been settled. Health Rising will keep you informed of future progress in this field.
Thanks to everyone who has brought us over a third of the way to our goal.
This was a fascinating and encouraging blog because it addressed a central question in long COVID (and ME/CFS, for that matter) that Health Rising has been following for as long as I can remember: is autoimmunity a real thing? The successful outcome of this study made it a delight to report that it looks like it is.
Your support has allowed HR to track this subject over the years. So thank you! And please support us in a manner that works for you. There’s much more to come…



This observation, autoantibodies to key receptors, fits with a hypothesis I have been working on for quite some time. I’ll split this post for ease of understanding the concept.
Part 1: ME/CFS and LC have a lot of ‘vicious circle’ properties, meaning you are ill today because you were ill yesterday. In engineering and system theory, a vicious circle is a self reinforcing feedback loop.
That is complex, so let me give a clear example we’ve all lived through: spreading of Covid19 during the pandemic.
There was a number that determined our public lives: the “reproduction number”. If it was bigger then 1, the pandemic was growing in number of infections per day. Then social measures against the pandemic got stricter. When the number was smaller then 1 the pandemic was shrinking and we could hope to soon retake part of our social lives.
This number actually was ‘reproduction number’ = ‘current number of newly detected infections’ / ‘previous number of detected infections’ with fixed (one week? not that important here) time intervals between the current infections numbers and the previous infection numbers.
In other words: if one week there were more new infections then previous week, the “reproduction number” was higher then 1 and the pandemic got worse. Roughly said: there was more virus in society.
The key thing for Part 1 is this: the same thing applies in our bodies. When infected with Covid19, we and our immune systems want to have an ‘inner reproductive number’ smaller then 1. I define an ‘inner reproductive number’ as ‘current load of virus in our body’ / ‘previous load of virus in our body’ with fixed time intervals between the current previous viral load numbers.
Part 2: When trying to get the pandemic under control, there was not one single way to do it. Simply killing all the virus worldwide at once was impossible. However many separate measures each did decrease the spread of the virus somewhat. On a state level, getting the “reproduction number” long enough under 1 was the goal in order to dampen the pandemic enough to get it under control.
Combining a lot of measures untill the reproduction number finally dropped was the task. Social distancing, waring masks, tracking people with active infections and isolating them, reducing time people spent in infected air (like having to shop in 15 minutes in and out), cleaning air through ventillation of rooms… all decreased how easily virus transferred and with it multiplied in society.
Now in our bodies, ‘simply killing the virus’ was not easy at all for many patients. Many failed and died. Other have so much scar tissue in their lungs they effectively lost half of their lung capacity for life.
This virus was pretty new to mankind. Many peoples immune systems didn’t seem to be able to ‘just kill it’ as their immune systems had no experience with dealing with it. This is a bit like science being unable to ‘simply remove’ all virus from society. So if that doesn’t work soon enough and the infection got dangerous, why would the body not follow the same approach the scientist trying to control the pandemic followed: trying what works to get the ‘inner reproduction number’ long enough below 1?
So what ‘measures’ could our bodies try out? Let me give some examples:
* producing lots of moderate amounts of ROS everywhere; enough to damage the virus so it’ll be less effective in reproducing but not too much in order to not kill our own healthy cells
*** block the receptors the virus uses to get entry to the cells; antibodies can do that ***
* increase blood clotting (even in absence of wounds); virus caught in a ‘ball of clotting’ will have few chances to get inside a cell to replicate
*** decrease blood flow; the slower blood flows, the longer virus has to travel from cell to cell if it travels through blood flow; then ROS and clotting will take out more virus before it can infect new cells; note: it’s a balance act as too strong constriction hampers the mobility of our own immune system too ***
*** decrease ‘metabolic speed’ of our own cells; cells in a state of strong decrease of metabolism will slow down the production of new proteins including viral proteins; looks a lot like inhibiting (Dauer!) our cellular machinery including enzyme, protein and ATP production ***
* …
A nice summary of what may have happened 🙂 – all in an effort to stop the infection
* producing lots of moderate amounts of ROS everywhere; enough to damage the virus so it’ll be less effective in reproducing but not too much in order to not kill our own healthy cells
*** block the receptors the virus uses to get entry to the cells; antibodies can do that ***
* increase blood clotting (even in absence of wounds); virus caught in a ‘ball of clotting’ will have few chances to get inside a cell to replicate
*** decrease blood flow; the slower blood flows, the longer virus has to travel from cell to cell if it travels through blood flow; then ROS and clotting will take out more virus before it can infect new cells; note: it’s a balance act as too strong constriction hampers the mobility of our own immune system too ***
*** decrease ‘metabolic speed’ of our own cells; cells in a state of strong decrease of metabolism will slow down the production of new proteins including viral proteins; looks a lot like inhibiting (Dauer!) our cellular machinery including enzyme, protein and ATP production ***
Part 3: Now I look at the four types of receptors Cort described as found increased in LC (I can’t read the paper due to pay-wall):
“Next, they wanted to see if they could identify subsets of LC patients who had distinct antibody profiles – and they found two of them. One was characterized by consistent positivity for beta-1 and beta-2 adrenergic receptors, endothelin A receptor, and muscarinic acetylcholine receptor M4.”
Note: I have no clue what the other subset is since the article is blocked behind a paywall.
When looking into it (not necessarily the best referrences; my time and energy is limited to dig deeper now) (many other receptor effects are available, I mention / choose those effects I feel lead to somewhere):
* endothelin A receptor (references follow my ‘line of thinking / thinking process’):
https://europepmc.org/article/PMC/PMC8155664, title “Antibodies Against Angiotensin II Receptor Type 1 and Endothelin A Receptor Are Associated With an Unfavorable COVID19 Disease Course.”
https://academic.oup.com/eurheartj/article/45/Supplement_1/ehae666.961/7835560?login=false, title “The role of angiotensin II and endothelin receptor antibodies in COVID19 patients with HFpEF”
https://www.sciencedirect.com/science/article/pii/S0022282822000517, title “Endothelin-1 is increased in the plasma of patients hospitalised with Covid-19”
https://www.ahajournals.org/doi/full/10.1161/01.cir.102.19.2434 saying “ET-1 acts through the activation of Gi-protein–coupled receptors. ETA receptors mediate vasoconstriction and cell proliferation, whereas ETB receptors are important for the clearance of ET-1, endothelial cell survival, the release of nitric oxide and prostacyclin, and the inhibition of ECE-1”
@researchers: ETB receptor agonist/antagonists…???
https://www.sciencedirect.com/science/article/abs/pii/0922410692901099?via%3Dihub, title “The selective endothelin ETA receptor antagonist BQ123 antagonizes endothelin-1-mediated mitogenesis”
=> ETA receptor modification has ***direct impact on mitogenesis via endothelin-1*** => direct impact on many LC sympthoms
* muscarinic acetylcholine receptor M4:
https://en.wikipedia.org/wiki/Muscarinic_acetylcholine_receptor_M4 saying “Activation of M4 receptors in the striatum inhibit D1-induced locomotor stimulation in mice. M4 receptor-deficient mice exhibit increased locomotor simulation in response to D1 agonists”
=> altering muscarinic acetylcholine receptor M4 messes with locomotion (movement) in the brain => direct impact on many LC symptoms
* beta-1 adrenergic receptors
https://en.wikipedia.org/wiki/Beta-1_adrenergic_receptor saying “Once these ligands bind, the ADRB-1 receptor activates several different signaling pathways and interactions. Some of the most well-known pathways are: … …cAMP activation of PKA: cAMP generated by adenylyl cyclase activates PKA”
* beta-2 adrenergic receptors
https://en.wikipedia.org/wiki/Beta-2_adrenergic_receptor saying ” whose signaling, via adenylate cyclase stimulation through trimeric Gs proteins, increases cAMP”
Now why do I zoom in on cyclic AMP (cAMP): because it plays major roles in cellular energy housholding. So I searched on:
https://onlinelibrary.wiley.com/doi/10.1002/jmv.28383, title “SARS-CoV-2 infection activates CREB/CBP in cellular cyclic AMP-dependent pathways” saying “In detail, cAMP-protein kinase (PKA) pathway has an essential role in SARS-CoV-2 infection, followed by the interaction between cyclic AMP response element binding protein (CREB) and CREB-binding protein (CBP) could be induced and leading to the enhancement of CREB/CBP transcriptional activity. The replication of Delta and Omicron BA.5 were inhibited by about 49.4% and 44.7% after knockdown of CREB and CBP with small interfering RNAs, respectively.” and “Taken together, our study demonstrated the importance of CREB/CBP induced by cAMP-PKA pathway during SARS-CoV-2 infection, and further provided a novel CREB/CBP interaction therapeutic drug targets for COVID-19.”
=> Now a full block of certain proteins is a ‘optimal research condition’ BUT that would inhibit Delta and Omicron BA.5 by about 49.4% and 44.7%. With ‘inhibit’ they mean “The replication of Delta and Omicron BA.5 were inhibited by about 49.4% and 44.7%”
=> A !!!single!!! measure, (crippling CREB/CBP through altering cAMP) could inhibit ***replication*** by (up to, when CREB/CBP fully clamped down) nearly 50%. I emphasis the word replication here. Why: it is *a single step in the chain of viral spread in our body*. And total ‘inner replication rate’ (in an important part) is the result of multiplication: ‘inner replication rate’ very roughly equals (percentage of virus able to enter cell) times (reproduction rate of virus) times (percentage of virus of surviving in transfer of blood) times…
=> slashing a single factor in a multiplication by (up to) 50% is like slashing the end result by p to 50%. It’s like having a single measure during the pandemic that could have slashed disastrous replication numbers of 1.5 to something well below 1! It’s a bit more nuanced, but still, it is a major part IF the body tried to overcome the virus while finding ZERO suitable antibodies against the virus.
* Now going back to muscarinic acetylcholine receptor M4:
https://www.mdpi.com/1424-8247/18/3/369 saying “M4 receptors belong to the metabotropic acetylcholine receptor family, linked to Gαi/o proteins. Activation of M4 by acetylcholine results in adenylyl cyclase inhibition, causing a reduction in cAMP levels.”
=> So 3 out of four receptors influence cAMP.
* back to endothelin A receptor
https://journals.physiology.org/doi/full/10.1152/ajprenal.00450.2005?doi=10.1152/ajprenal.00450.2005, title “Endothelin-1 stimulates NO production and inhibits cAMP accumulation in rat inner medullary collecting duct through independent pathways”
=> So 4 out of 4 receptors found in this study have a major effect on cAMP AND cAMP has a major effect of Covid19 replication rates!!!
=> This shows quite a picture of these receptors being *altered* (auto antibodies effect their working but not all need to do so in the same way. I can very easily imagine the body can produce antibodies that are agonists, antagonist or just block the receptors by sitting in the way depending on the needs) IN ORDER to reduce viral reproduction.
=> In addition, they also tend to alter blood flow. If that alteration means reducing, then it leaves the virus travelling slow in more narrow tubes with more blood clotting to catch it and ROS to take there time to damage and remove intact virus.
=> So altering the working of these receptors has the (IMO strong) POTENTIAL (not proven it does in this specific case) to both slash replication rates as the amount of replicated virus that survives well enough to be able to infect other cells. That alone can slash viral load *over time*.
=> We seem to have an ‘alternative immune system’ at work here that hampers the spreading of the virus by attacking it at key phases (reproduction rates, survival rates, removal rates, defect rates…) without the need of ‘the classic immune move of tagging virus and removing it byt immune cells. It is an alternative way to use the immune system without ‘tagging’ a single virus with antibodies. It is a POTENTIAL alternative if finding in time a suitable antibody against the virus fails.
=> By what I see, it is not a behavior by accident but a reasonable well coordinated behavior of the immune system and cell behavior at large. It is part of our makeup *as a fallback if most everything else fails*.
=> This system, by its very nature, would and should act slower up to a lot slower then the ‘classic’ immune system. We see that clearly in Covid where even plenty of patients not ending up in LC tend to take time (weeks up to months) in order to get rid of symptoms (potential side effects of the immune system sort of deliberately messing with both energy production and blood flow in order to contain the infection ‘via the method of getting the inner reproduction number long enough below 1’.
=> Note that it is not “classic adaptive immune system” or “alternative immune control”. Both can and will coexist. The quicker the body finds antibodies against the virus and the better the quality of them, the lest the alternative will be used” and the quicker the recovery will go.
Hi Cort, A question: if I remember correctly, the autoimmune substances found (Scheibenbogen et al.) in the subgroup of ME/POTS patients are natural autoimmune substances that also occur in healthy people, but in lower quantities. I haven’t read the entire article on Long COVID that you describe. Does this also apply to Long COVID patients? Or were unique, specific autoimmune substances found there?
Great question. The answer is yes! 🙂 I looked it up. They are both. We all have “naturally broad repertoire of low‑affinity, often polyreactive “natural autoantibodies,” that affect these receptors. So they’re sitting around, not doing much, and not very targeted.
The idea is that when the system is put under stress (inflammation/infection) the natural autoantibodies can be “refined” into pathogenic ones. They become more highly targeted (“class switched” and begin attacking the receptors. You can tell that they’re pathogenic if as their levels rise the symptoms get worse – which is, I believe, what this study showed.
You can’t tell which way they’re going by looking at them but certain features (isotype, subclass, epitope, affinity) can reliably indicate they’ve become pathogenic.
Who knew! Thanks for the question – it cleared that up for me. 🙂
The elephant in the room, though: does autoimmunity play a role in something as neurologically complex as PEM?
Absolutely I think it could. Here’s my laymen’s scenario. Exercise triggers autoantibodies to act up which damages the microcirculation and cause low oxygen conditions to to occur. When the exercise is over the oxygenated blood returns to now damaged, low oxygen capillaries. That actually causes MORE damage. PEM results because the body now needs to try to clean up the damage and with the repair mechanisms not working well, that takes more time.
One question, though, is what is causing the inadequate response to exercise in the first place and then the inadequate repair mechanisms that cause/contribute to PEM. Why don’t they click in? Why is the response to exercise so underpowered?
I don’t think we know and what roles autoantibodies play is an open question I think. Conceivably, since they can knock out any part of the body, they could be hitting energy production, etc.
The authors think more things are in play though. We shall see! 🙂
Interesting. I will read the paper in due course.
Are we talking a relatively small subset (25-30%) or a much bigger one?
It depends on how many people fit the neurological profile. I suspect that most people do. Regarding that specific autoantibody subsets which affects the adrenergic receptors, I don’t think the paper says, but other studies suggest its from about 25-40% of ME/CFS patients if I remember correctly. I’m sure that number will be refined more over time.
2 years ago AMA Update | American Medical Association
Akiko Iwasaki on what causes long COVID, brain fog, the Yale Paxlovid study and long COVID treatment
https://www.youtube.com/watch?v=fEjwilGWApg
And:
Long COVID 2025: Symptoms, diagnosis, post-COVID treatments and the latest long COVID research
Akiko Iwasaki, PhD,
“Common symptoms of long COVID vary widely. Some patients primarily experience neurocognitive problems, while others may have cardiovascular or gastrointestinal symptoms.
We also found evidence of hormonal changes, including lower cortisol levels and differences in sex hormones. For example, some female patients showed lower testosterone levels.
This complexity makes diagnosis and treatment challenging. We are working to develop better biomarkers that can classify patients into different endotypes, with the goal of improving diagnosis and enabling more targeted treatments.”
https://www.youtube.com/watch?v=ilIOh4cZiNI&t=16s
Yes, that’s what they’re doing – trying to identify biomarkers and subsets. Hopefully they will plunge into autoantibody discovery. I imagine they will.
Thanks!
Cort, imagine if every patient were systematically tested for a broad panel of autoantibodies and their symptoms, disease course, and treatment responses were reported to a centralized research center.
How much faster could biomarkers and biologically meaningful subsets be identified? Instead of researchers working with small, fragmented cohorts, they could analyze data from thousands of patients, making it much easier to detect patterns, validate biomarkers, and distinguish distinct disease mechanisms.
It seems like discoveries that currently take years—or even decades—could potentially be accelerated dramatically.
I was hesitant for a long time to post my proposal.
https://swaresearch.blogspot.com/2026/06/proposal-standardized-autoimmune-and.html
However, with millions of people continuing to suffer from autoimmune and post-viral illnesses while progress toward accurate diagnosis, standardized testing, and effective treatment remains far too slow, I can no longer justify remaining silent.
The need for coordinated action is urgent. Delayed diagnoses, inconsistent testing protocols, and inadequate reporting systems continue to leave many patients without answers, appropriate care, or recognition of the true scope of these conditions.
If you agree with the content and goals of this proposal, I urge you to share it widely and forward it to healthcare professionals, researchers, policymakers, patient advocacy groups, and others who can help advance this initiative. Meaningful change will require collective awareness, engagement, and action.
Any high systemic toxicity can cause pericyte damage: viral illness, high blood sugar, chemical, poison…. Perhaps someone can clarify for me how the study establishs the difference between a responsive post viral pericyte clean up operation verses faulty autoimmunity. I’m not educated in such matters.
It is interesting, & I’ve wondered about that for awhile. That said, I also wonder if autoimmunity is part of it, how those who got sick after physical trauma fits with this. I did only read the gist, though – not up for more, so there maybe something I missed.
I’m not sure but the pericyte findings came, if I remember correctly, when they determined which tissues the antibodies from the long COVID patients reacted to or attacked. That suggests to me that this wasn’t about cleaning up the pericytes so much as the long COVID patients antibodies actually attacking them.
I’m thinking more about my original question re autoimmunity & people who got sick following physical trauma…..and maybe it has to do with the cell danger response turning things down &/or off after said trauma, which could include affecting the mitochondria…and we know the mitochondria imapct the immune system, so perhaps that’s the connection….just a thought.
I’ve been trying to look more into the possible connection between physical trauma, mitochondria, & possible autoimmune issues, & found that mitochondria damaged by physical trauma can promote inflammation. Researchers are linking this type of mitochondria damage (& resultant inflammation) to autoimmunity. So, that’s another possible way that ME that was triggered by physical trauma could also be an autoimmune subset.
part 4: short summary
(@Cort part 3 in moderation)
* The blog mentions 4 receptors one LC subgroup has increased auto antibodies against
* These receptors all 4 happen to have a clear and strong influence on cAMP levels
* Research found cAMP levels have a criticial role on Covid19 viral replication rates
=> That and a number of other things (IMO strongly) indicates that *in these subcases of LC* our immune system tries to find (during active infection) antibodies to part of our own cells that significantly reduce the ability of the virus to replicate
in addition, ‘messing’ with these receptors has the potential to:
* decrease blood flow
* increase oxidative stress
=> That has the ability to damage (due to ROS) and isolate (in blood clots) virus more, reducing how much of the replicated virus effectively infects new cells
In simple math: ‘amount of newly infected cells’ is proportial to ‘replication rate of virus once inside cells’ times ‘virus being undamaged’ times ‘virus being free enough (not in clots) to get into cell’
so when for example each factor is reduced by 10%,
we go from 100% * 100% * 100% = 100% to 90% * 90% * 90% = 73%
=> Even small individual impacts do reduce viral reproduction a lot. Having multiple ways to slash replication compounds this effect.
=> Soon, you get a situation where each 100 infected cells are no longer able to infect a 100 new cells, and the infection runs out of steam and dies away. This is much like when 100 people no longer infected 100 new people in the Covid19 pandemic. When that was the case, scientist said the replication number was below 1 and the pandemic was contracting and easing.
This way of fighting a pandemic however took a lot of time as we all experienced. And so does this ‘alternative way’ of using the immune system.
The alternative here is that the immune system targets parts of the own cells that will slash reproduction rates of the virus rather then targetting the virus directly. Why? Likely because it fails to find a suitable strong antibody (to the virus) in time.
In reality, it will be a combination of both the classic way (immune system directly targetting the virus) and this hypothesized alternative way (immune system targetting select key parts in our cells in order to slash succesfull infection rates of uninfected cells).
This behavior aligns with reovery from LC being a near continuous spectrum. It ranges from very quick recovery (those who find high quality antibodies quickly) over those who recover but it taking weeks or months (having a mix of both immune modes) to those ending up in LC (those having poor anti-virus antibodies and more alternative immune functionning).
This idea is adapted to this LC subgroup, but variants on this idea for ME and other LC subgroups exist.
Now why do the people in the LC with this subgroup not recover? Many IMO will no longer have virus in them, even if some will. But body wide low grade inflammation (due to these auto antibodies) alone could wrongly signal the body that it must produce more of these auto antibodies. I am working on that too but that is for another time.
” But body wide low grade inflammation (due to these auto antibodies) alone could wrongly signal the body that it must produce more of these auto antibodies.” that’s the vicious circle isn’t it? Autoantibodies cause tissue damage – which sparks an immune response – and more autoantibodies (!) and perhaps different ones. People may have/probably have a particular series of autoantibodies – which is one this situation is more complex that others where one autoantibody dominates.
Let me try and ‘correct’ this answer with the one you gave in the comment above in “Cort Johnson on June 4, 2026 at 9:41 am”.
“We all have “naturally broad repertoire of low‑affinity, often polyreactive “natural autoantibodies,” that affect these receptors. So they’re sitting around, not doing much, and not very targeted.”
and from the same “Cort” comment comming from the paper I can’t read due to paywall:
“The idea is that when the system is put under stress (inflammation/infection) the natural autoantibodies can be “refined” into pathogenic ones. They become more highly targeted (“class switched” and begin attacking the receptors.”
If I use this and combine it with my hypothesis of
A) the body seeking to target (mulitple) specific parts of cells that help slow down viral spread and replication
B) the body prefering to spread out side effects and inflammation due to A) by doing these adaptations body wide so that the immune system won’t / (almost) can’t go into a ‘focused, local spot’ attack mode
THEN nearly the only way to ‘adapt’ key signalling routes (to try and work around a big problem like fierce acute infection) *while using auto-antibodies for it in a desperate way* is to target key signal proteins or parts of the body that are truely abundant around the body.
A close second is trying to find auto-antibodies to receptors or places of entry the pathogen uses to enter cells BUT even then the specific blocked part should be abundant around the body in order to try and prevent runaway auto-immune disease developping
=> blocking key receptors that regulate viral reproduction speed, viral ease of moving (bloodflow…) and amount of oxidative damage are ‘ideal’ candidates. Just like the ones this study found.
Now comes your part Cort:
“”We all have “naturally broad repertoire of low‑affinity, often polyreactive “natural autoantibodies,” that affect these receptors. So they’re sitting around, not doing much, and not very targeted.”
=> So they sort of seem to be ‘waiting’ to be activated in all healthy and less healthy people. And the body has a dedicated mechanism to do it as you further wrote:
“the natural autoantibodies can be “refined” into pathogenic ones. They become more highly targeted (“class switched” and begin attacking the receptors.”
Now the classic view is that this class switching is only for turning antibodies against a *specefic* pathogen active if it is ever detected again and that doing so for auto-antibodies is a ‘defect’ and troublesome part.
With a system engineer, I dare say this looks *very very* well like a natural *function* of the combination ‘having these antibodies ready to be jacked up in all healthy people’ and having ‘a very quick switch to turn the *desired* / *needed* ones active on the fly’.
There are even hints (need to find proper links) that plenty (near all?) people do this during acute infection. Difference is they are quickly able to undo this during the recovery phase.
Another hint is that doctors start to use artificial man-made auto-antibodies to actually *treat* diseases (I found many links). It would surprise me nature hasn’t learned the trick before them.
In this idea, ‘hitting the gut first’ during an infection (as is observed!) is a feature rather then an unhappy defect. Gut lining has plenty of surface area and a very high recovery / cell renewal rate. And epithelial blood vessels follow behind that. They’ll provide plenty of ways to difuse / dilute the auto-immune reaction and IMO can convert danger of quick auto-immune burning holes in *very hard to repair* organs into slower *spread* low grade inflammation.
I dare say this all (plus some more, but later on that) combined makes so much sense from a system point of view that it may be near spot on (for an auto-immune central long-type subgroup).
I don’t know if this relates, but I think so. In March, I tripped over a trailer hitch and fell hard on the concrete driveway. I fractured my wrist and bruised my hip and knee on that side. When I got back the results of my quarterly blood tests, I had a “10” for inflammation (normal is around 3) and positives for all ANA markers (and my doctor runs a bunch of them). So I am wondering if my body’s reaction to this injury resulted in these out of range markers rather than the markers being indicative of autoimmune disease. It will be interesting to see if the numbers have changed when I get my next blood work.
Sorry to hear this. I hope you heal well.
Thanks for sharing. It does relate:
https://pmc.ncbi.nlm.nih.gov/articles/PMC7616895/, title “Complex Autoantibody Responses Occur Following Moderate to Severe Traumatic Brain Injury”
=> Note: many patients’ FM (or ME/CFS) starts with an accident with whiplash or equivallent
not only injury induces it, also “chronic life stress”. https://www.nature.com/articles/s41380-020-0672-1, title “Multiple inducers and novel roles of autoantibodies against the obligatory NMDAR subunit NR1: a translational study from chronic life stress to brain injury”
Interesting from that paper: “NMDAR1-AB are induced by chronic life stress and exert antidepressive effects in mice and human.”
=> NMDA receptor auto-antibodies protect against depression. Note that depression rates in ME/CFS are remarkably low despite often very poor life quality.
and quotes “Chronic life stress in humans: replication of our previous findings of enhanced NMDAR1-AB seroprevalence in young migrants” and “Novel antidepressive, ketamine-like role of NMDAR1-AB upon access to the brain in humans and mice”
another paper https://www.sciencedirect.com/science/article/abs/pii/S0306452214007325, title “Autoantibodies in traumatic brain injury and central nervous system trauma”
quotes “An overproduction of autoantibodies is observed in CNS injury.” and “As discussed, the activation of immune mechanisms post CNS trauma involves the disruption of the BBB with exposure of brain-specific proteins (potential antigens) to the immune system along with break down products (BDPs). The breach in the BBB allows the self antigen/immune cell interaction to occur activating the immune response post TBI (Diamond et al., 2013). ”
@Cort, @Betty: Another interesting paper is https://www.sciencedirect.com/science/article/abs/pii/S1568997223001209, title “Functional autoantibodies: Definition, mechanisms, origin and contributions to autoimmune and non-autoimmune disorders”
quote “Here, we do not only explain functional autoantibodies but also summarize the mechanisms underlying the effect of such autoantibodies including receptor activation or blockade, induction of receptor internalization, neutralization of ligands or other soluble extracellular antigens, and disruption of protein-protein interactions. In addition, in this review article we discuss potential triggers of production of functional autoantibodies, including infections, immune deficiency and tumor development.”
=> It explains many things regarding the types of auto-antibodies, what triggers them and the many ways they can alter health and signalling without needing immune cells to attack the affected cells. It also is in line with my idea of “if the body wanted to develop auto-antibodies against receptors in order to try and influence cell signalling in a desired way, there will be a variety of potential auto-antibodies possible that fullfill the expected role wether up, down or other regulation is needed”.
I wish I could be a part of a clinical study. I have FM before the pandemic, but I caught a “mild case” but have lost taste and smell for 2 1/2 years and many times each time I was in contact. The last time I got it it almost put me in the hospital, not because of respiratory or cardiovascular issues, but because it had me so disoriented. I have also had severe cognitive decline and brain fog. I used to be a teacher and now I can’t really even hold a conversation some days. My quality of life has really suffered and doctors can’t tell me squat (I’m in Kansas and there is no one hear who will even see me for LC). I want my life back!
So sorry to hear Melissa. It’s particularly hard when your brain was your pride and joy and you loved explaining things to others.
I imagine that you are strongly (very strongly!) in the neurological subset and that at some point they will be able to identify the autoantibodies that are particularly associated with high levels of brain fog. They must be attacking certain parts of the brain.
It does help me when I feel low that I remember that so many formerly capable people are dealing with the same situation.
Thank you for the validation. I hope they can figure things out sooner than later.
Hopefully if CAR-T Therapy works for more people with autoimmune diseases as it did in that woman who recently recovered from lupus (she also had two other autoimmune diseases) and still remains in remission 14 months later of all 3. It may help in all autoimmunity.
I remember saying to a friend years ago, if they could beat the root cause of autoimmunity in one disease, they may be able to beat all autoimmune diseases. Now that’s looking quite promising.
I personally have long suspected autoimmunity in ME/CFS, more so because of the high amount of close relations on my mother’s side who all have autoimmune diseases (at least five that I know of ) and a cousin of my mother has a severe crippling fatiguing illness that he got in his 40s and he’s now in his 80s (incompetent psychiatrists tried to blame that on mental illness).
Also because flu vaccinations caused so much severe and permanent worsening to my condition I wonder if those vaccinations somehow excited the autoimmune response.
I’m not anti-vax though, as they have helped millions of people worldwide. It’s just a significant minority of us unfortunately can’t have them due to ME/CFS worsening.
I have also read that some people with autoimmune diseases were having more adverse reactions to vaccines than healthy controls. Mainly a worsening of their autoimmune symptoms.
I wonder if vaccine issues in 1 in 5 ME/CFS patients (and possibly LC patients too) could possibly be loosely linked to these subgroups who also may have an autoimmune version of ME/CFS. Maybe that’s a too simplistic view, but if there’s anyone who understands immunology and ME/CFS I’d be interested to hear their opinion.
I see the recent study (in the blog above) on long covid emphasizes a much broader autoantibody profile against neural, vascular, and other proteins compared to the two Swedish studies published back around 2020 that found >>Autoantibodies to beta-adrenergic and muscarinic cholinergic receptors<> AI summary : Common symptoms that could theoretically result from if someone had high levels of autoantibodies targeting β-adrenergic and muscarinic receptors, symptoms would depend on which receptors were affected and whether the antibodies were activating, blocking, or disrupting their function:
* Rapid heart rate (tachycardia)
* Palpitations
* Blood pressure instability
* Dizziness when standing
* Orthostatic intolerance
* Fainting or near-fainting
* Exercise intolerance
* Abnormal sweating
* Temperature regulation problems
* Gastrointestinal symptoms (nausea, bloating, constipation, diarrhoea)
* Dry eyes or dry mouth (especially if muscarinic receptors are blocked)
* Bladder dysfunction
* Sleep disturbance
* Tremor
* Anxiety-like physical sensations
* Fatigue
* Brain fog and concentration difficulties <>Swedish autoantibody ME/CFS (2 x cohorts)
https://www.sciencedirect.com/science/article/pii/S2666354620300727
>>CAR-T Cell Therapy Achieves Complete Remission in Woman with Three Simultaneous Autoimmune Diseases – MedPath Trial<<
https://trial.medpath.com/news/car-t-cell-therapy-achieves-complete-remission-in-woman-with-three-simultaneous-autoimmune-diseases?utm
For some reason part of my text disappeared
I wanted to say just before the AI summary…
“Just take a minute to think about what those specific autoantibodies are targeting! if you had high levels hammering away at those essential receptors that are involved in running the autonomic nervous system, which regulates heart rate, blood pressure, blood vessel tone, digestion, sweating, pupil size, bladder function, and many other automatic body processes.
So I fed the question into AI (without mentioning ME/CFS ) to see what symptoms would arise. And no surprises many are ME/CFS and long Covid symptoms…”
I just hope that if as an autoimmune disease that permanent damage has already been done. For instance if rheumatoid arthritis is treated early, the joint damage is not reparable. Does the report mention anything about that?
Edits — ….damage has not already been done
…..Early, the damage is reparable
I think when it comes to ME/CFS, it’s not actually structural parts of the body that have been attacked, like the myelin sheath in MS, joint cartilage in rheumatoid arthritis, or pancreatic cells in type 1 diabetes.
I think if we could reverse the autoimmunity, we would see cells regenerate. However, if most of those cells are gone, they may not be able to rebuild the structure completely. for example, not rebuild the pancreas or the sheath surrounding nerves. Although I think some of regeneration would still occur.
If ME/CFS is an autoimmune disease and it’s instead targeting receptors on cells, then I’m not as worried about regeneration, because the next generation of cells that develop should come back with the receptors that were previously being attacked. Or maybe the receptors on existing cells can just start doing their job again when not being bombarded buy autoantibodies.
For example I’ve had ME/CFS for 36 years. The first five years were pretty bad, nearly a year bedridden. the next five showed slow improvement, and the following 15 years I was constantly in crash and recovery, a bit like the stock market up and down but slowly overtime going up.
Until I got to the point where I was nearly fully recovered. In fact, I was working full-time and going out drinking and dancing.
I even worked on building sites although I did have to take afternoon rests and I did sleep much longer than other people. But I had my life back and it was fantastic!
Unfortunately, a flu vaccination 10 years ago triggered the entire disease all over again, and I ended up permanently severe. I still remain severe today. (Several years prior also noticed feeling quite unwell in autumn but I thought it was to do with the seasons, but when we started looking at the medical files we could see a correlation with the flu shot and 7 to 10 days later me going through a worsening period.)
But that story near full recovery should be enough to help people see that substantial recoveries do happen (even though I later fell back down the hole). What I mean is that there didn’t seem to be permanent damage. I still had a some brain fog, more to do with that visual haze, but it the cognitive issues quite closely tracked the severity of the disease. Severe ME/CFS gave me severe brain fog. Very mild ME/CFS gave me mild brain fog.
So I don’t think much permanent damage was done, because if ME/CFS is autoimmune, it may be affecting cellular function by targeting receptors with autoantibodies. (Remove that attack and those receptors will be able to function again)
Rather than attacking and destroying structures. That’s my take on it. I may be wrong, but hopefully that helps
Thank you! I hope you’re right; certainly sounds plausible. Your illness progression would certainly support the theory.
Thanks for replying.
When an immune cell finds an (auto-)antibody bound to our cells, it IMO makes few difference if it is bound to receptors or other parts of the tissue. It is an incentive to clear up the cell.
That said, if the auto-antibody is bound to a receptor on the surface of the cell, there is chance that many of them get cleared at a slow but constant rate. It is normal for many receptors to be ‘swallowed’ when triggered by something but it is not a given. It depends on many variables.
That way, the density of the number of bound anti-bodies to the cell decreases and with it the chance for immune cells trying to clear the cell. Increased inflammation will likely still be there, but this IMO should dampen the most aggressive forms of the immune system trying to clear the cells.
I think lower density of receptors per surface area are also reached by the (I think) relatively modest amount of receptor auto-antibodies produced and more so by huge number of receptors available to bind to. In that way, the density of auto-antibodies attached to cells is strongly reduced.
Think of 1 ‘bag’ of auto-antibodies to the thyroid being produced by the immune cells in the body per day. As the thyroid is tiny in volume, they’ll bind at very high concentration in that tiny volume. That’ll be a red flag for immune cells to engage fierce action in that small volume.
Now think of this same amount of 1 ‘bag’ of auto-antibodies being produced against blood vessels. It doesn’t even need to be receptors on blood vessels. Since the volume and surface area of blood vessels in the body is way larger than the volume of the thyroid, they’ll be spread way more across the body. With it, local reaction to targeted cells will / should be way more moderate. Chances are that we’ll have increased inflammation here versus damaging auto-immunity in the thyroid case.
If it are auto-antibodies to epithelial receptors in general rather then blood vessel receptors, then the produced auto-antibodies will be further spread out. Sure, more of them will likely be produced but IMO often not proportional to the number of potential targets (especially if the immune system is ‘inhibited’). The local densities of bound auto-antibodies to receptors should further decrease and with it local immune reactivity.
Here, the four mentioned types of receptors targeted by auto-antibodies are common to not only all epithelial cells but even other cells, leading to more spreading.
So where inflammation of the epithelial gut lining and (granted a bit worse) brain-blood-barrier sounds as a bad thing to happen, it may be just as well a tool to spread out and transform auto-immunity to one particular spot / organ and ‘transform’ it into spread out body-wide low grade of inflammation. Transforming potentially crippling and constantly degrading auto-immune disease to very inhibiting strong inflammation and being out of any energy for daily activities of life.
To answer the final question: yes, I think most of the damage is reversible but very hard to get there.
Very simply said:
Use high pressure water jet cleaning to uniformally clean all of a big concrete wall in one hour, and it will be cleaner.
Use the same high pressure water jet cleaner a full hour on a single spot of the same concrete wall and you probably make a hole in it.
With auto-antibodies to types of receptors that near all cells have, the immune cells need to spread their action out. This makes it more like the ‘cleaning entire wall’ case. Use auto-antibodies specific to only a small part of the spinal cord, eg to myelin in MS, and you’ll probably burn holes in it.
Because there are complete recoveries (several of which involve brain retraining – suggesting that the damage is not permanent and is more due to “crossed wires” in at least some people) and because some researchers think ME/CFS is more than anything a “signaling” problem, I think you’re on the right track.
I hadn’t heard it expressed that way before. Thanks for the ideas 🙂
Will need to come back and fully read when I’m better.
For now, in this small (n= 20) 2025 study https://pubmed.ncbi.nlm.nih.gov/39759581/ “Efficacy of repeated immunoadsorption in patients with post-COVID myalgic encephalomyelitis/chronic fatigue syndrome and elevated β2-adrenergic receptor autoantibodies: a prospective cohort study” , Scheibenbogen and colleagues gave immunoadsorption treatment to a cohort of patients who all had been shown to have beta2 adrenergic receptor antibodies, and 70% of them improved from repeated immunoadsorption treatment (not cured, but marked improvement as far as I understand?). If I remember correctly from a Scheibenbogen presentation, it seems that being positive for beta2 adrenergic receptor antibodies could therefore be a kind of predictor that, with a 70% chance of success, immunoadsorption might help you.
A question for you, Cort: You mention sensitivity to odors – is this from your interpretation of what the affected brain areas do? Or is sensitivity to odors specifically mentioned in the study?`(Can’t look it up because of paywall).
I’m always keen to read about findings re. odor sensitivity as it is among my most disabling symptoms. Thank you!
I don’t remember exactly what the paper said but that’s what those parts of the brain do. I was really struck about the impact to the sensory assessing areas of the brain. First of all, the antibodies appear to be attacking them, but they also attack the parts of the brain which makes the symptoms feel worse. Not a great combination.
Daniel Clauw thinks these diseases are primarily sensory diseases. I’m sure more is going on but I like his thinking focusing on how the brain processes sensory stimuli.
Thank you, Cort!
Endothelin receptors, adrenergic receptors and muscarinic receptors seem to have 1 thing in common: blockade of them seems to be able to slow a broad class of cancers. References see below.
Now this has quite some implications. The large scale Rituximab trial failed, while the smaller but still fairly large trial of Fluge and Mella was rather promising.
As per referred papers below and more (too tired now, very late here), blocking endothelin, adrenergic and muscarinic receptors show good potential to slow or even prevent cancer development.
As per my *hypothesis* spread in this blog, *deliberate* production of autoantibodies to receptors of our own cells *might* be used by our bodies as a tool to try and overcome very difficult challenges the body otherwise fails to overcome (so a desperate attempt).
Now ALL patients in Fluge and Mella’s original study were former cancer patients (that is where the observation came from as their ME/CFS improved after Rituximab treatment). Plenty of the later larger scale trial by them have a good chance to also have cancer related issues as they all are patients who were brought in the study by Fluge and Mella. In the biggest scale trial, ME/CFS patients were selected from the total ME/CFS patient group and this study failed.
So: per my *hypothesis*: if the body gets more resilient to plenty sorts of cancer when Endothelin receptors, adrenergic receptors and muscarinic receptors are blocked, chances would be high to find plenty of patients whos immune system developped autoantibodies to block these exact receptors. And Rituximab exhaust B-cells, the ‘production factories’ of antibodies including autoantibodies.
=> According to my hypothesis the, the percentage of ME/CFS patients with increased (and problematic for ME/CFS symptoms) endothelin, adrenergic and muscarinic autoantibodies should be far larger among (former) cancer patients then the average ME/CFS patient population.
=> Therefore, the Rituximab test results should be a lot better when ME/CFS patients were recruited by cancer experts among their own patients. And bombing their autoantibody producing B-cells should have far greater chance to relieve their ME/CFS symptoms!
Note: 2016 paper https://www.sciencedirect.com/science/article/pii/S0889159115300209 with title “Antibodies to β adrenergic and muscarinic cholinergic receptors in patients with Chronic Fatigue Syndrome” from Scheibenbogen et all found 3 out of those 4 receptors increased in ME/CFS too!
=> This also is another indication that the body is likely able to produce autoantibodies in response to severe problems as a tool to try and control disease since autoantibodies to these receptors have potential to slow a large class of cancers and per this comment chances IMO are high that the group of ‘ME/CFS patients that have some relation to cancer diagnosing’ are likely those with increased chance of autoantibodies to these receptors.
=> IF this hypothesis would pan out, then there is likely quite an overlap between receptor autoantibody dominant ME/CFS and LC fatigue / exhaustion and post cancer fatigue. And if any field has plenty of cash for research, it is the cancer field…
References cancer versus receptor antibodies:
https://www.mdpi.com/2072-6694/14/9/2322, title “Muscarinic Receptors Associated with Cancer”
quote:
“In conclusion, there is a correlation between epidermal growth factor receptors and cholinergic muscarinic receptors, survival clinical differences adjusted by stage factor, and an association between gene expression and immune infiltration level in breast, lung, stomach, colon, liver, prostate, and glioblastoma human cancers. Thus, targeting mAChRs appears to be an attractive therapeutic alternative”
Note of mine: the paper says that in some cancers the muscarinic receptors are *overexpressed* and that worsens cancer. So there the muscarinic receptors are VALID AND CORRECT targets of the (auto-)immune system.
https://pmc.ncbi.nlm.nih.gov/articles/PMC3534394/, title “The roles of beta-adrenergic receptors in tumorigenesis and the possible use of beta-adrenergic blockers for cancer treatment: possible genetic and cell-signaling mechanisms”
quote:
“Conclusion
Beta-adrenergic blockade may play a role in the prevention and treatment of cancer… …Further investigation of the relationship between β-adrenergic antagonists and cancer is required.”
https://advanced.onlinelibrary.wiley.com/doi/full/10.1002/adtp.202000289, title “Endothelin Inhibition Potentiates Cancer Immunotherapy Revealing Mechanical Biomarkers Predictive of Response”
https://journals.physiology.org/doi/full/10.1152/ajpregu.00532.2015, title “Endothelin therapeutics in cancer: Where are we?”
quote:
“Supportive evidence for the involvement of ET-1 in tumor progression and metastasis comes from different studies.”
and
“Levels of ET-1 are higher in patients with muscle-invasive bladder cancers, which are prone to metastasize and which correlate with reduced patient survival, thus indicating ET-1 could be a potential biomarker for lung metastasis”
=> So here again, some cancers *show* / correlate with increased ET-1 (holds ET-a from the ME/CFS study as subclass) AND increase risk for metastasis (spreading of cancer to other organs).
=> In those cases (increased ET-1 and active cancer), ET-1 (including Endothelian A) are VALID AND CORRECT targets of the (auto-)immune system
https://www.mdpi.com/2227-9059/12/3/511, title “Endothelin-1 and Its Role in Cancer and Potential Therapeutic Opportunities”
quote:
“Many preclinical efforts have been made to target ET-1 expression within cancer, such as by using ET-1 receptor antagonists, many of which have been approved for treating pulmonary hypertension. Targeting ET-1 has been shown to improve the response to various other cancer therapeutics, highlighting the potential benefits targeting this peptide may exert”
https://aacrjournals.org/cancerres/article/81/13_Supplement/1776/667645/Abstract-1776-Tumor-derived-endothelins-regulate, title “Abstract 1776: Tumor-derived endothelins regulate antitumor immune responses through macrophage endothelin B receptor”
Note of me: this paper discusses endothelin B, not endothelin A but endothelin B has a role in regulating endothelin A levels.
Don’t worry guys, a few sessions of CBT will cure these antibodies /sarcasm
oh yeah…and throw in some graded exercise for good measure….a little more sarcasm.
Grateful for the finer minds who have broken this study down for us here and given us their
own insights. Keep the faith everyone. We are inching ever closer …..