


Part 1 introduced how Cortene became involved in chronic fatigue syndrome (ME/CFS), and introduced its hypothesis that a maladaptation within the limbic system, which shapes our response to stress, may underlie ME/CFS.
In Part II Health Rising takes a deeper dive into nuts and bolts under girding Cortene’s hypothesis.(Given the long nature of this post you may want to print the blog out using the print/PDF buttons on the bottom left hand side of the post)
Stress
“Stress” is an unfortunate term. Usually we think of stress as emotional; in biology, though, stress means any threat that disrupts the balance (or homeostasis) of the body. The stress response or HPA axis, prepares the body to respond to the threat. Any threat then, whether infectious, emotional, physical, chemical, etc, will initiate the stress response.
Once triggered, the stress response suppresses non-critical functions such as growth and metabolism (i.e., hypothyroidism, long linked to ME/CFS) and reproduction (i.e., hypogonadism, also connected with ME/CFS). It also releases cortisol to make sure the brain, heart and muscles have sufficient glucose (at the expense of less critical functions like digestion). Cortisol also primes the immune system for action (and has a delayed proinflammatory effect).
As noted in Part 1, studies indicate that chronic stress causes a progression from high to low cortisol and can result in the development of cortisol sensitivity – a situation in which the body becomes more responsive to cortisol. (When cortisol sensitivity occurs low cortisol can have the same or greater effects than high cortisol does in healthy individuals.) This increased cortisol sensitivity cannot be measured by cortisol/synacthen tests (which measure level not effect) but it does results from epigenetic changes that can be shown.


Cortene’s new hypothesis for ME/CFS will shortly be tested in a small exploratory drug trial
Studies indicate that ME/CFS patients show the same alterations in cortisol levels and cortisol sensitivity seen in chronic stress. These findings help to explain the overlap of immune and metabolic symptoms found in ME/CFS and chronic stress but they do not explain the neurological issues found in ME/CFS.
The stress response also involves brain neurotransmitters such as serotonin, norepinephrine, dopamine and GABA (gamma-aminobutyric acid), which focus on and deal with the stress in a stressor-specific way. These neurotransmitters – which may have been under-appreciated in ME/CFS research – are at the core of Pereira’s hypothesis.
Animal studies indicate that short, medium and long term responses to stress are governed by two factors, CRF (corticotropin-releasing factor) and UCN1, that affect the release of serotonin (and norepinephrine) in the brain and cortisol (and epinephrine) from the adrenal glands.
If these two factors do indeed govern the response to stress in humans, Pereira/Cortene believe that if they can get at the switch controlling them they can reset the stress response system. That can be achieved they believe by altering the receptors found on the neurons that govern the stress response.
Some background…
The Players
Corticotropin-releasing factor (CRF or CRH) is a hormone produced in response to stress that initiates and shapes the stress response. It has two receptors it can bind to on neurons: CRF1 and CRF2.
Receptors are proteins on the surface of a cell which make it possible for the cell to respond to its environment. When molecules lock onto receptors they trigger actions in the cells –such as the production of hormones, neurotransmitters, cytokines, etc.
Cells don’t just respond to their environment, however. By altering the kind and number of receptors on their surface they determine the kinds of response that are possible.
As we’ll see, cells often prime themselves for one type of action by loading their surface with one type of receptor. In the scenario below, different levels of stress; i.e. different levels of CRF, have very different effects on two receptors found on stress response neurons.
Depending on how much of the stress hormone CRF is present one or another receptor will dominate the surface of our stress response neurons. Each of those receptors, in turn, will have very different effects on how much serotonin those neurons will produce. Keep your eye on serotonin in the following scenario
Low Stress States (CRF1) – Low levels of stress (low CRF) activate CRF1 receptors on GABA-releasing neurons of the raphe nuclei and limbic system, triggering the release of GABA, which decreases serotonin release in the limbic system. During low levels of stress, then, CRF acts via CRF1 to inhibit serotonin. (The CRF1 receptor, then, inhibits serotonin.)
High Stress States (CRF2) – High levels of stress (high CRF), cause the CRF1 receptors to leave the surface (or internalize) on GABA-releasing neurons and bring CRF2 receptors to the surface of serotonin-releasing neurons. During intense stress, CRF acts via CRF2 to pound out serotonin. (CRF2, then, increases serotonin.)
Urocortin 1 (UCN1) – UCN1 is a peptide that interacts with CRF1 and CRF2. When the stress dissipates, UCN1 causes the CRF2 receptors to internalize in the serotonin-releasing neurons, and the system returns to baseline. UCN1, then, normalizes the stress response.
A Dysfunctional System Appears
Step I: Serotonin Release
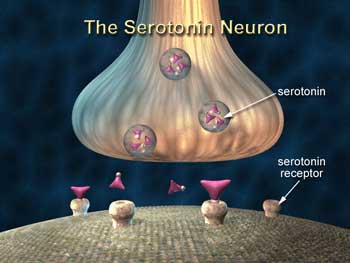
Serotonin producing neurons and serotonin play key roles in Peireira’s hypothesis.
Serotonin is the bogeyman in this hypothesis. In the brain, serotonin is produced by the raphe nuclei in the brain stem, and serotonin neurons extend throughout the limbic system (including the hypothalamus) and the prefrontal cortex, affecting all the other neurotransmitters and coordinating the response to stress.
Step II: Desensitization
The raphe nuclei and limbic system shape the stress response (by incorporating assessments of risk, reward, history, etc), but under intense stress they can desensitize the 5HT1A autoreceptors that normally halt the stress response – allowing it to run amok. Pereira/Cortene believe this is what is happening in ME/CFS.
No stimulatory part of the body is ever designed to be “on” all the time. Because stimulating any system for too long will cause it to break down, any stimulating response comes equipped with brakes. The brakes on an out-of-control serotonin response in the brain are the 5HT1A serotonin autoreceptors. (“Auto” meaning that when these receptors sense serotonin around a particular neuron, they reduce that neuron’s release of serotonin).
Studies in both animals and ME/CFS patients suggest that riding the serotonin stress-response system for too long causes the 5HT1A “brake” to fail and the 5HT1A autoreceptors become desensitized.
With that the foundation of this hypothesis is complete.
Step III: Chronic Fatigue Syndrome (ME/CFS) Begins
In situations of intense stress (e.g., infection, trauma, emotional distress), high levels of CRF in the raphe nuclei (and limbic system) propel serotonin promoting CRF2 receptors to the surface of serotonin producing neurons. The high levels of serotonin produced cause the 5HT1A “brake” to fail. Once that happens, high serotonin levels prevent the release of UCN1 and the re-establishment of homeostasis.
UCN1, remember, causes the serotonin promoting CRF2 receptors to disappear back into the neuron. With UCN1 unable to return the system to normality, the CRF2 receptors remain on the neuron’s surface – telling it to keep pumping out serotonin.
With the neurons (in the raphe nuclei and limbic system) now packed with serotonin-producing receptors (CRF2), and the brakes on serotonin release gone (desensitized 5HT1A autoreceptors), the HPA axis has become sensitized. Now even minor stressors, like exercise or emotional stress, or even stimulation (light, sound, conversation) can initiate a major stress response. This is what Pereira/Cortene believe is happening in ME/CFS (and probably in related diseases such as fibromyalgia).

Peireira’s hypothesis proposes a way even minor stressors can produced major stress reactions in the body in ME/CFS
Animal studies demonstrate how this progression occurs. Intense or prolonged stress (particularly early in life) causes CRF2 receptors to remain on the surface of the neurons long after the triggering stress has gone causing 5HT1A desensitization and behavioral issues (impaired memory and learning ability, anxiety, PTSD-like behavior).
There is hope for these animals, however. Removing the serotonin promoting CRF2 receptors (via sophisticated experimental techniques) eliminates the 5HT1A desensitization and the behavioral issues.
Consequences of Excess Serotonin
If ME/CFS patients’ brains have been turned into serotonin pumping machines, what causes the immense fatigue and post-exertional malaise found in this disease?
It turns out that serotonin plays a vital role in the motor cortex and spinal cord as well. At low levels, it increases motor neuron excitability, making the muscles more responsive. As activity increases, serotonin levels increase, and the motor neurons/muscles become even more responsive.
However, when serotonin levels become too high, they inhibit motor neuron signals, preventing muscle contraction (to avoid muscle damage). Several studies suggest that reduced motor cortex excitability, motor preparation, motor performance and central activation during exercise may be present in ME/CFS.
Pereira/Cortene believe that the hypersensitive serotonin response in ME/CFS patients causes them to reach this threshold very quickly. Intriguingly, their hypothesis also may illuminate an unusual experience that many people with ME/CFS may feel: that their muscles feel more like they’re stuck or paralyzed than that they’ve have run out of energy – and that any stimulation can make the situation worse.
Increases in serotonin have been directly implicated in increased pain sensations, cognitive dysfunction, migraine, sensitivities (light, sound, etc.), sleep dysfunction and depersonalization. Indirectly, by stimulating other neurotransmitters such as dopamine and norepinephrine, serotonin regulates everything from behavior (e.g., mood, perception, reward, anger, aggression, attention, appetite, memory, sexuality) to physiology (e.g., gastrointestinal functioning, blood coagulation, blood pressure, heart rate).
But why have these postulated CRF2/CRF1 maladaptations not shown up in the ME/CFS research to date? Tests of blood and other bodily fluids will not pick up a problem existing only on specific neurons in the brain. Nor can the serotonin output of these specific neurons be measured. While cerebrospinal fluid can get close, it lacks the precision to identify a problem involving a tiny subset of neurons in the brain. Biopsies of specific neurons in the raphe nuclei and limbic system in the brain are needed.
Some indirect evidence does suggest that this hypothesis may be correct. The limbic system neurons postulated by Pereira/Cortene to be problematic are, in fact, the same brain regions that light up in Nakatomi’s ME/CFS work. Cleare’s 2005 pet scan study indicates 5HT1A autoreceptors are desensitized across the entire limbic system in ME/CFS; and Maes has found antibodies to serotonin in ME/CFS patients’ blood.
Of course, there’s more to ME/CFS than altered brain chemicals.
How does this hypothesis account for the hypometabolism seen by Naviaux, Armstrong, Fluge and Mella and Davis?
The Pereira/Cortene hypothesis proposes that a sensitized HPA axis causes even small stresses (including the disease itself) to release cortisol into a system with increased cortisol sensitivity. The resulting excess of cortisol stimulation creates insulin resistance, a situation where the normal insulin-directed uptake of glucose by cells is inhibited by cortisol.

Peireira believes his hypothesis could account for the low energy states studies have found.
The result is increased blood levels of both insulin and glucose, shown to be present in ME/CFS patients (Armstrong/McGregor). Elevations in insulin and glucose in ME/CFS serum could also explain Ron Davis’s work, in which healthy cells become abnormal in ME/CFS serum but remain normal with the addition of pyruvate (which reduces insulin resistance in animals). The resulting lack of glucose in ME/CFS cells could also explain why these cells appear to be using non-glucose substrates (Naviaux, Fluge and Mella).
What about the Immune System?
The notion of a sensitized HPA axis could also help explain the immune dysfunction found in ME/CFS. Cortisol has both an initial anti-inflammatory effect and a delayed pro-inflammatory effect which is intended to deal with any injury arising from the stress. In chronic stress, however, excessive cortisol stimulation suppresses Th1 (cellular immunity) and promotes Th2 (humoral immunity), with attendant changes in cytokines. This could explain why ME/CFS patients are prone to both allergies (driven by increased Th2) and opportunistic infections (driven by decreased Th1). Note also, that under certain circumstances, a corticosteroid (which is essentially a cortisol mimic) could worsen an ongoing stress response and worsen ME/CFS.
Other Symptoms?
A sensitized HPA axis would also result in excess norepinephrine (NE) release potentially explaining many of the autonomic symptoms found in ME/CFS, such as a high resting heart rate, low heart rate variability, low blood pressure and orthostatic intolerance, and abnormalities of the gastrointestinal system (potentially leading to leaky gut) and urinary system.
What about the four facets of the disease (mentioned in Part 1)?
Diverse triggers: Any stress, e.g., infection, emotional distress, physical trauma, chemicals, could result in the postulated CRF2/CRF1 maladaptations. A sudden outbreak of ME/CFS would likely be due to a single pathogen sweeping an area. Plus, Hickie found in the Dubbo studies that different infectious triggers cause post-viral fatigue in ~11% of cases. The fact that those with the most intense immune response (i.e., greatest symptoms) tended to come down with ME/CFS, suggests that for the known infectious triggers, an underlying natural, likely genetic, proclivity to the development of ME/CFS exists (see below).
Sudden/gradual onset: Over time accumulated CRF2/CRF1 maladaptations could result in a sensitized HPA axis. This could happen via an intense stress sufficient to produce maladaptation (sudden onset), or the accumulation of sub-threshold stresses over time (gradual onset).
Gender bias: The female stress response releases more CRF than does the male, has more CRF1 and CRF2 receptors overall, has greater concentrations of CRF1 and CRF2 in certain parts of the limbic system, and shows reduced rates of CRF1 and CRF2 internalization. Other differences exist as well, but all point in a direction that heightens the female response to stress. Importantly, many of these differences emerge at puberty, which explains why the gender bias in ME/CFS is only evident post-puberty (i.e., no gender difference pre-puberty).
Symptom range and variability: Animal work has shown that stress-based adaptations are stressor-specific and result in different maladaptations in the limbic system. This suggests that the symptom presentation of a given ME/CFS patient depends on that patient’s cumulative stress history. This could explain how some patients can present with primarily physical symptoms while others have primarily cognitive symptoms. The symptoms vary over time because the level of the HPA axis response is affected by whatever stress the patient might be under at the time of measurement. This explains why some patients are able to reduce their symptoms with meditation and other calming techniques while some are not. It also explains why putting patients under stress (e.g., via CPET or cardiopulmonary exercise testing) improves the consistency of study results in ME/CFS.
Other Evidence
The risks of developing ME/CFS, and increased symptom severity after one has ME/CFS, have been associated with a wide range of genes involved in the HPA axis and serotonin. That list includes CRF2, CRF1, TPH2 (involved in serotonin synthesis), 5HTT (the serotonin transporter), NR3C1 (the cortisol receptor), POMC (a precursor for ACTH, which triggers the release of cortisol under stress), and TH and COMT (both involved in dopamine, epinephrine and norepinephrine synthesis/breakdown).
Note that all of these genes are involved in the stress response. Defects in them could either make it difficult to unwind or tamp down an ongoing stress response or result in a hypersensitive stress response. Either of those situations could lead to the loss of the 5HT1A “brake” described in Pereira’s hypothesis, and potentially explain the Dubbo findings.

Genetic studies suggest Cortene may be looking in the right area
Finally, Peireira’s hypothesis has something to say regarding the mixed effects SSRI antidepressants have on people with ME/CFS. Neurons communicate via neurotransmitters (such as serotonin) jumping the gap between them. SSRIs work by blocking the serotonin transporter (a channel in the neuron that recycles serotonin in the gap). This was proposed to solve depression by increasing what was thought to be low serotonin.
In fact, depression is caused by high serotonin releases from specific neurons. Blocking the serotonin transporter, causes serotonin to build up outside these neurons until, 2-8 weeks later, it triggers 5HT1A auto-regulation which reduces these neurons’ release of serotonin.
The trouble is that SSRIs block the serotonin transporters on all neurons. This means SSRI will eventually reduce serotonin from neurons releasing excess serotonin (via auto-regulation), but it will also increase serotonin release everywhere else (just not to a level that triggers auto-regulation). This is what causes the many side-effects of the SSRIs—and, notably, these are the symptoms of ME/CFS.
This long overview suggests that Pereira/Cortene’s hypothesis could explain much of what’s happening in ME/CFS. But is there evidence that turning down CRF2 receptor activity as they propose could work?
In Part 3 we’ll dig into that question and describe the upcoming clinical trial.
- The Cortene Way: New Drug to Be Trialed in Chronic Fatigue Syndrome (ME/CFS) Soon – Pt. I
- Cortene III – A New Drug for Chronic Fatigue Syndrome (ME/CFS): The Clinical Trial
- Cortene IV – The Cortene Chronic Fatigue Syndrome (ME/CFS) Drug Trial Begins



Excellent work. Really hoping for this to pan out.
Fantastic research data. Im Hopeful for a cure soon!
What research data? There isn’t any yet.
Thanks – me too! 🙂
Thanks Cort! Great to look at yhings from a different angle. And how lovely of them to name it after you!!
?
I know! I hope it works out! 🙂 🙂
Any thoughts around changing from SSRI treatment for anxiety/depression for people with Chronic Fatigue to MAOI’s? Which are enjoying something of a comeback.
Soon with stage 3 cort please. My daughter has asked me to look for it? Please God
This sounds very promising! Despite it all, we feel at least lucky that my girlfriends ME is not 20 years ago, now a days there is real hope.
My self I’m ailing from anxiety, and had been for about 15 years.
Could there be an application of cortene in treating anxiety/depression as well? Bells are going off in my mind with all this about serotonin, norepinephrine, 5ht1a etc? Is there any way to get this question to the scientists?
“However, when serotonin levels become too high, they inhibit motor neuron signals, preventing muscle contraction (to avoid muscle damage)”. I wonder if that’s what happening to cause the pain in fibromyalgia? In addition to CFS/ME I also have fibromyalgia and I’ve noticed when I’m fighting any infection my muscles get so taut they are more like bricks than elastic fibre. When the infection clears up, the muscles aren’t nearly as painful anymore.
EC, my dad and I have ME/CFS (and my dad’s sis). It obviously has some genetic ties. Autoimmunity can run w/ it. A family study Dr. Salahi is doing out of Univ of Albany NY might reveal some fascinating info regarding illnesses common in families w/ ME/CFS and ME/CFS in multiple family members in some cases. Anyway, my dad and I both get terribly tight muscles at times of stress (viral infection, emotional stress that’s prolonged). Interesting you mention that. My shoulders are the worst, and TMJ. Occasionally my chest gets knots and that’s been scary at times w/ chest/ribs not expanding well. My legs get tight occasionally if I’m at my worst. My dad gets similar but his legs and sciatica areas are his worst area when they act up. Followed by shoulders/neck which are a more common but a little less severe issue for him. And his chest at times. I’m very interested to have a genetic screen and find out if the genes mentioned that pertain to stress are mutated or something in us. Bad copies perhaps? I know I was put on an SNRI,Pristiq (a serotonin norepinephrine re-uptake inhibitor), when I had bad body pain w/ a total relapse of ME/CFS after a partial recovery for years. It helped the pain (CNS depressant) but then I felt like I was going to die! It was horrible! First, it made me not get deep sleep. I am in the minority w/ ME in that I usually fall asleep fast and I sleep deep the majority of the time…but at my sickest it’s not restorative. Even though I felt I was in a deep-sleep-coma. When I could sleep on the med I woke in the night and could hardly breathe my nervous system was so suppressed. Something with brain chemicals and sensitization to the neurotransmitters. After only taking it a few days and going off it, I had bouts for 3 weeks every couple days waking up like that. I was scared to go to sleep and this is when I needed sleep most. Then the final straw, I woke up with one of these episodes so bad it scared me that I might die. I want to get help to my parents bedroom where my dad was and 1/2 way down the hall my legs froze. Then worked again. IDK if my brain was 1/2 stuck in sleep or what.
My shoulders, neck and arm are really bad too at times. JT, Interesting you mentioned sleep. For the first few years of cfs/me, I never got into a really deep sleep. Then about 20 months ago I started a multi-level Buddhist-style meditation program and now there are times when I sleep so deeply it’s hard to wake up. For me it hasn’t been alarming because I had been so deep-sleep deprived for many years. But I also tend to drop off into a sleep anytime I sit down and get still, and I’m out like a light, totally unaware of anything (that probably just happens because of cfs/me not the meditation). I agree there may be a genetic component as my daughter is showing some similar signs (brain fog, rheumatoid arthritis, etc). I too have had chest tightness, and almost always have some kind of upper respiratory infection at the same time.
Most serotonin are made in the gut!
The serotonin made in the gut does NOT cross the blood brain barrier into the brain so that’s neither here nor there. The gut serotonin is used to control intestinal contractions that move the digested food along.
“However, when serotonin levels become too high, they inhibit motor neuron signals, preventing muscle contraction (to avoid muscle damage)”
I’ve remarked this sentence and it’s role in the whole hypothesis too.
However to me it feels like quite a stretch. We have in general too ???few??? muscle contraction? ME and FM people report tense muscles times and again. Many Physical Therapists say to their ME/FM patients that they can’t relax their muscles but rather put them in a continuous state of contraction.
Isn’t it more accurate to say that ME/FM patients muscles are so often “pre-contracted” that their muscles both are not able to contract much further and are weakened by this continuous effort the contraction requires? If so, what resembles “being unable to contract muscles” would be “unable to relax muscles sufficiently so that muscles can do work by going from relaxed to contracted state”.
If indeed our muscles would be pre-contracted most of the time, it may conflict with this muscle/serotonin/contraction part of this hypothesis. I’d still very much like their drug to work out well for us though, but I’ve seen too much promising things go wrong to put all my hope on it. Thanks and good luck to the researchers for looking into ME/FM!
Thank you Cort for all you do. This now explains why my PEM can include a 2 day jaunt in bed with absolutely no ability to rise. Any attempt at movement makes it obvious that my muscles are not listening to my brain at all. No discomfort but a bit scary for sure.
Cort, will we know if cortene works or not before the end of this year?
We should know if Cortene is safe and effective in a small number of patients in a couple of months. If Cortene passes its first test then determining if it is effective in ME/CFS overall could – if everything works out – and we know how often that happens – take as little as 1-2 years.
Fibromyalgia patients should probably watch the Cortene trial closely as much of what’s going on in ME/CFS in these areas could apply to FM as well.
So far, it’s ticking all my boxes. I’ve recently been able to add panic attacks to my list of “wtf?”s. From the reading that I’ve been able to retain, that’s hyper-arousal of fight/flight response, now on a hair trigger.
If Cortene works out (and I so very much hope it does), it will be a life changer.
Me too, Erika. Have had ME for 18 years but for the past five I’ve been trying to work out why I feel like I have PTSD.
This post explains a lot, Cort. And 1-2 years for a possible release date? That’s just amazing!
I’m wondering if it works the other way as well…I have ‘fight or flight’ issues from childhood trauma (check out http://www.acestoohigh.com), and am now wondering if that could have triggered my ME/CFS? I don’t remember not being tired, even back into grade school. How crazy would that be if my PTSD/fight or flight syndrome started the CFS?
Sounds like a great hypothesis. It would be so wonderful if the drug worked!!!
This makes so much sense- and fits with everything I have experienced. I am really hopeful that this will lead to treatment. Thanks for all you do Cort- you give us hope.
This is some complicated stuff. Are you allowed to say- on a scale of 1 to 10, How confident are they about this drug? 10 being most confident.
And what if there’s another extremely stressful event? Would it throw things off again?
No, because it resets the dysfunctional part that’s overreacting to stressors in the first place. Wouldn’t that be nice.
Amazing report, thanks, Cort.
“One to two years,” each like dog years, seven years in FM pain years.?
A race between a Cure and the Reaper for some of us
Yes, indeed. We don’t want to get ahead of ourselves and be in for a let down but If this drug works out in the exploratory trial – a very big if! – Cortene hopes that the FDA will put the drug on fast-track status accelerating its testing schedule. It would be surprising if it did not as Ampligen, I believe, was put on a fast-track status and the FDA has acknowledged that ME/CFS is a very serious disease with many unmet needs.
Please hang in there.
No kidding, more like 8ME yrs! I am 64 1/2 (got ME at 24) and my life has little value or meaning anymore, as I am the worst, for the longest, that I have ever been (thanks to fibro taking away my remissions). But better late than never, if I make it that long.
~ Personal note only to Caramia ~
Dear Caramia,
I just wanted to send a note of caring to you. I’m hearing that things are at the lowest point ever … “the worst, for the longest, that you have ever been.” I pray that since you wrote this on Feb. 18th, there has been some little ray of Light. Please know that even though we have never met in person, I (a fellow ME and FM sufferer) am with you in spirit.
At 62, my ME has been “only” twelve years, compared to the 40 LONG YRS (approx. 14,600 DAYS) you have endured. I profoundly admire the great courage you have had in just living each day. Those without ME + FM don’t see what a real heroism lies in just getting through day by day … just staying in life … just bearing the losses and inner feelings resulting from our devastating, often homebound/ isolating, usually financially-ruining, and definitely life-robbing illnesses. But I KNOW you are an unseen hero over 40 long years! Years that, as you say, feel each like 8 years.
I’m also writing you because I feel we have in common the feeling that our lives have little value or meaning anymore, and even your phrase “if I make it that long.” You have prompted me to think about why I feel the same way … so I will share my reasons in the possibility that you might in turn share yours … and in so doing we might gain meaning on a similar journey.
1. I used to define my value and meaning esp. through the altruistic “called” work I loved to do and which absorbed my attention — and through volunteering during vacation tim (my esp. loving helping in Asian refugee camps). Both stopped with my becoming bedridden (all 12 years) with my ME, FM (and sleep apnea + 3 types of severe mostly nerve pain). I did such a wide variety of work that It seems even some close friends don’t know what I did … which makes my life lose meaning, since all I gave in those 30-some years does feel like my greatest value to this world and others.
2. I yearn to travel overseas again (often) — and sometimes think I still can — but as soon as I have an after-exertion crash, I can easily let it go. As you probably know well: when one is so sick that it takes all one has just to get through one day, — a “hoped for” life dream (something beyond one’s exertion limitations even on good days, if one’s honest) becomes SO unrealistic, so inconceivable, that there’s no emotion to abandon it. On those days one sees the reality of one’s severe debilitation with such clarity.
3. My life had meaning in wanting to live a caring, spiritually growing life with “Mr. Right,” — and I had a # of proposals from wonderful beaus but who were not quite “the one,” + a short engagement with U.S. Congressman who became verbally & emotionally abusive. Finally with ME I had a beau (who I’d known since I was 10!) but was too sick to even go on dates, participate in occasions with his family, help around the house, and do even a small % of all he did to so lovingly help and take care of me (that included 2 hrs of daily commuting!) He ended up burning out. This gave me a vivid picture of how romantic relationships would hence be lop-sided, my needing care, my limiting so much of what we could, — so I have had to let this lifelong expectation go (believing no one would want to be with me, anyway, because of my looks’ change from all the weight gained after lifelong slenderness. I think this loss has a much more profound effect on me than I even want to acknowledge.
4. So I am single, childless …. live alone …. and am very lonely. My very beloved Mom and Dad passed away in 2016 and 2013. My sister died suddenly in 2010. My remaining sibling lives 4 hrs away and has mostly treated me horribly since around the time of my ME diagnosis (though this may be changing.) His and my sister’s children are all nephews, not as close as one step-niece sometimes is … than girls tend to be. This family absence, now, affects my feeling of not being of value. I can compare it to the very meaningful life I DID have through ’16 while doing a lot every day to love, brighten the life of, and take care of Mom — in her last years with Alheimers. (In some ways, then, she was even like the child I never had had.)
5. Mostly from the church I used to be close to, a group of friends calls themselves my “spiritual family” (mostly just coming to an annual birthday party, and one group Workday I had) — and 3 of them my “Executive Committee” (for extra support, doing needed tasks to help me and helping me discern decisions). I couldn’t be more deeply grateful for them, — especially the one woman who is single and sometimes comes to stay in my guest room & watch DVDs & support me, and the “click” I have with one (happily married) handsome man friend (whose wife I love) whose energy is so special, supportive, caring … who I think allows me to positively channel the part of me who likes being with men but in a way that is non-stressful. Still, I don’t feel I have any friend who I can “go through life with,” as in share housing and grow old together platonically: I believe this would add value.
6. Another life of value, for me, would a form of housing I can envision that would be in a huge old Victorian — or interesting new natural wood house — full of bedroom suites, to share with single of like-values — where growing old til the end together (with shared assistants and health care aides) we’d be set up as a live-in community/foundation (or raising money) with a mission to plan educational offerings: films, workshops etc about social justice and spiritual growth and global forms of community and indigenous leaders/traditions and so forth… with action avenues … (and with games nights, crafts, fun films + theme gatherings thrown on the side.) But I am too exhausted to catalyze almost anything now, esp. something way way big like this.
7. Anything that needs to get done can pretty much only be done by people I hire or a rare volunteer. I also have such an unstable life without helpers who have stayed long-term and being too exhausted to even be able to find another when one leaves, then taking ANYone who comes along, only to find they have health problems or other problems that make them leave — a vicious circle. This has meant continuously not moving my life forward, the same goals never getting accomplished. I cannot put in words how depressed this has made me … how truly desperate for good long-term helpers. I feel If I really could secure this — after a lot of orienting, and surely some % of guiding, and after all the tasks I would still have to do to move life forward, I could sell my house, move to some inexpensive peopled, fulfilling, like-valued somewhere, where I feel safe, — and at last maybe have some freed up time with some energy. (I have hope for more energy also b/c I start w/ the esteemed Dr. Silvieri this July, so have hope I may gain some energy). Then I would have the question: what could I spend time/energy on that would make me want to live life and feel meaning in it, make me feel I am of value, and that I left some things of value/meaning by life’s end.
8. Despite my memory of names and places starting to falter, I’ve thought of maybe a book of vignettes from my life of service — with the lesson learned, or meaning in that encounter or moment, or snippet of humble wisdom — that I could at least leave for my nephews and their descendants.
(I feel for you in this regard, maybe not having had a chance for years of working/volunteering … though you possibly mentioned remissions.). Or …. well … I have plenty of time to think of other ideas. For now I just want to be in better touch w/ my nephews, and be the most ethical and loving person I can be.
Please pardon how lengthy my thoughts have been on the commonality I felt we had from your post. If you would be so inclined to share your feelings on the subject, I would be an attentive caring reader, and we could support one another in some way. Please remember, dear Caramia, that — even though we have not met in person — I am with you in spirit.
With warm wishes,
Alden L. from Washington DC
What a beautiful, wonderful post Alden. Good luck with Dr. Sivieri. I hear that he’s very good. I love your idea of shared housing.
Thank you so much for sharing. I’m eager to learn more (and volunteer myself as guinea pig at the mere chance of recovery!).
Ohmygosh, sign me up now. I would be a guinea pig for this trial in a heartbeat – what do we have to lose, after all??
If it is approved the cost wouldn’t be affordable to 80% of us unfortunately. I’ve spent everything I had on this disease even Ampligen…..I’d like to think differently but it’s reality for me. I hope it’s a success but if you can’t afford it what’s the use……
Why do you say that it won’t be affordable?
F
I just read that Ampligen would cost over $40,000 a year. And when is a new drug not expensive?
John, but Ampligen (though VERY helpful and effective for some) doesn’t have FDA approval. If this (Cortene) was approved by FDA (and it already has phase 1 animal and human safety data, thank God!), it would mean it’s covered by insurance most likely. So, it should be more affordable than some things. It’s terrible that Ampligen is still pay out of pocket due to lack of FDA approval. It is very helpful for a subset w/ low NK cell function, chronic viral issues. And who don’t have an autoantibody block to the TLR3 receptors.
Drug makers will give a brand drug to you for free, in their patient assistance program. They generally approve people, I believe, as the govmt reimburses them. Perhaps someone with more knowledge can speak to this. I have been approved for all PA programs I have applied for, and I have exceeded income, etc. Although the sheer number of ME pats may break the system!
I must add that I applied to 2 programs for brand name drugs I couldn’t afford and they turned me down.
To me this hypothesis makes more sense for fibromyalgia than it does for Myalgic Encephalomyelitis. It seems like it better explains those symptoms which do include fatigue but also a greater focus on pain and sensitization. To me it doesn’t explain the energy / metabolism differences that occur during PEM (see Workwell research) as well as say the intracellular sodium hypothesis. However, it would be interesting to see if there is some way that the intracellular sodium issues cause stress to trigger this as it would explain why fibro is commorbid to so many MECFS patients.
Great article! This is really promising. Does Pereira think that cortisol is the molecule that Ron Davis filters out of patients’ blood with the effect of ending the metabolic problems?
Cortisol is relatively small IMO, and Ron Davis is aiming at a relatively big molecule to filter out IMO. If I recall both correct then the answer is likely no.
Thank you Cort for pulling this all together and presenting it in a we/I can understand (at least with the help of my wonderful husband). After so many many years of struggling against my mind and body, and searching for answers – I am inspired by this hypothesis. I do hope I live long enough to see a successful treatment for this debilitating condition.
I wonder whether other bodily dis-functions could be explained by this hypothesis. My body seems to do a poor job of regulating blood pressure and body temperature. Usually my blood pressure is high not low, though I’ve never checked it when I’m bedridden. I’ve experienced bad reactions and side effects from all of the medications my doc has prescribed so I don’t medicate for it. Another weirdness is that I seem to randomly flip flop between hot/sweaty and cold/clammy. Back and forth especially at evening and at night.
Rough stuff Leslie – my guess is that those symptoms are not uncommon in ME/CFS. Don’t they just sound like a hypersensitive system that isn’t sure how to regulate itself? To me that could fit with this hypothesis.
Cort, interesting stuff. So if that serotonin hypothesis was true, would that mean that SSRI’s would make our disease worse? (I know you touched on this in the article, but I didn’t quite understand it)
Serotonin and SSRI’s are very complicated. Some people with ME/CFS are helped by them and some are not. You’d need an expert to really assess that relationship.
Leslie I also run hot/cold. The hot is usually at night and almost always correlated with an upper respiratory infection. It’s hard for me to always know when I have a uri since I tend to have chronic sinusitis plus allergies and I hardly ever run a fever since contracting cfs/me. I’ve recently started having blood pressure fluctuations after a lifetime of routine 110/75-80 but we think it’s caused from thyroid rx and issues.
I hope Part 3 will discuss the results of Cortene’s animal trials.
Wow Cort a nice hypothesis but scientifically I do not see any objective measuring possibilities (only proof of concept), ‘the brake’ is defective. That’s how it feels. As if the accelerator pedal is pressed until you are completely exhausted. But we still don’t know what causes this effect it is all speculation.But they are looking at the right place now! Many times I wrote that the stress system was defective that usually results in angry reactions from me patients this is what ME is.ME is an abnormal stress response to every stimulus.
The mechanisms of aspirin‘s effect was only recently shown. Yet it worked fine before that. Drug trials are not really scientific research illustrating mechanisms but rather just test effects. I very much hope this drug has the desired effect.
Please look at my “stress-adaptation hypothesis of chronic fatigue syndrome” in the journal “Fatigue: Biomedicine, Health & Behavior” (I am a psychiatrist who has conducted in-depth interviews with several thousands of CFS/ME patients).
Dear professor van Houdehove, The difference between your thinking and mine is that in my vision there is a physical defect in the stress system. Your vision is that it is maintained by emotional causes (fear etc..). Correct me if I’m wrong.
Sorry Houdenhove….
In my experience of recovering from CFS it’s caused by unconscious stress.. which is caused by blocked emotions.. when you tap into the blocked emotions, you heal.. symptoms of CFS are simply emotions with the volume turned up… an exaggerated stress response caused by blocked emotion..
yes, I was a patient of yours in Pellenberg belgium, the psychosomatic revalidation. And I only get worse! We lyed with several patients on one room and no one got better. Afterwards, if I contacted them, we all had a trauma from how we were treated by you and your staf. Thanks a lot!
ps I had later by an endocrinologist a synachtentest and my glands did not work by themself but they did when they get stimulated by the test.
A friend of mine has the same.
ps I think there are many things going wrong in our brain, even neurotransmitters but I do noy buy this hypothesis as the root of our illness. I have much more hope on ron davis, omf and the researchers they work with, stanford, nancy klimas, and it is much mor complex then what is written here. I gp for like ron davis sais multiple personalised tests and multiple personalised treatments. if this hypothesesis is true and can help 1% , ofcource I will be glad
Gijs, it seems to be looking in at least one of the right areas, I agree. Not sure if it’s why downstream things go wrong. When I first got ME it was right after puberty and w/ viral infections. And when I had a total relapse/re-onset it was a ton of stress and not fighting viruses well. I distinctly remember feeling (most heightened time of this feeling) tremendously physiologically stressed…to where I had to have calm and couldn’t physically handle outside stress or upset. My glucose metabolism was also off I know b/c I just felt horrible after eating much of any sugary food at my sickest esp. It still makes me feel crappy. But at my sickest I didn’t even eat carrots or fruit. Made me feel bad. It was wonderful when I could enjoy chocolate ice cream again! (I know am dairy free except butter and occasionally mozzarella b/c low on whey). I also was yellowish around my mouth at both times. Worst w/ teen illness before partial recovery. Possibly that goes w/ liver/gallbladder stuff related to a study Dr. Maureen Hansen’s group just put out. I also think w/ the stress response locked on, we don’t heal good and sleep isn’t restorative. When I had a total relapse I had acne (had since Jr high) and it wasn’t healing. Like I’d sleep, wake up, and healing hadn’t occurred. It just got worse and I got sicker til total crash.
I am also yellowish around the mouth. I guess I missed the Hanson gallbladder information- could you please point me to it? Thanks!
No Rebecca you’re example is not he way (Stress respons) ME works and what i mean with abnormal stress reaction. That is mentally.
Many thanks Cort for bringing this to our attention – it all sounds so completely true… I hope it does pan out to be so.
Hi all, I have started a thread to track any serotonin, or other therapies we feel may work if this theory is correct. I would welcome anyone’s thoughts on this. Apparently activated charcoal can reduce serotonin. Not a permanent fix, but potentially very helpful!
https://www.healthrising.org/forums/threads/cortene-study-what-to-do-in-the-meantime.5851/
I wonder how this meshes or if it could mesh w/ RCCX gene module theory by Dr. Sharon M…something. RCCX theory seems to mesh w/ Naviaux’s work (Cell Danger Theory). I also wonder about all these things (theories, Cortene) and the issue of EDS-hypermobility type. EDS-HT exists in a significant subset of ME patients, myself and my father included. Dr. Peter Rowe sees these patients in a pediatric clinic. I know blood which contains immune cells is a connective tissue, and if EDS-HT is a defect in connective tissue…it could effect immune function. Maybe these issues are downstream effects of gene alterations due to a maladaptive stress response? Or partially the issue could be that people just happen to have hypermobility that have ME and and it becomes pronounced due to deconditioning from ME. Otherwise healthy, active people also have EDS-HT. Thoughts?
I’m sorry if my comment is too simplistic. Brain fog and pain are really bad today.
I have ME and fibro – but I’m also Bipolar. I’ve been on SSRIs for over 20 years, but only fell ill 12 years ago. Before I became ill I felt great, tons of energy. Now I’m ill, and I don’t know what I would do if I went off of my SSRIs after being on them for so long.
Another thought, doctors are so quick to tell ME/CFS patients that they are “just depressed” and then give them SSRIs. In your opinion, is that just making things 100x worse? If so, that would be gross malpractice!
Last thought, why are chronic depression symptoms so very similar to CFS? What’s the mechanism there that makes them so alike that it’s hard to tell the difference?
Because depression is also a stress-related disease. The causes are the same. The mystery is why some people get CFS and others simply have depression.
Also, depressed people don’t get PEM from activity – socializing, working out (low or high intensity both) have a significant positive impact on depression, and should be encouraged for good reasons. For M.E. pasients it’s the exact opposite, getting severe PEM and a flare of other symptoms kicking in after abnormally low intensity and increasing exponentially from higher activity – regardless of how positive your mindset is and whether or not you expect the PEM to occur. It’s maybe the easiest single cursor to distinguish M.E. from (most) other diseases, although of course you need to look at the rest of the picture too.
That the psychiatric wing ignores this is to me baffling. But I guess when you’re schooled a certain way your confirmation bias makes you want to see and recognize your field of expertise in everything, therefore fixating unproportionally on the things that overlap, giving less heed to the rest.
Well said…
Terrific post and hypothesis Cort and an amazing number of boxes ticked off for ME/CFS. I hadn’t known we have insulin resistance issues in ME/CFS, which makes sense of my newest symptoms too. Their trials will be so informative to follow!
I’ve been seeing a similar pattern in research for type 1 diabetes, inflammatory bowel disease, MS, other autoimmune diseases and other illnesses as proposed above (altered nervous system responses to stress, immune system affected and issues with Th1 and Th2 etc etc). The studies suggest that the disease(s) we develop are influenced by three underlying factors:
1) genetic predisposition, as you mentioned, for some of us though not everyone
2) timing of exposure early in life, especially prenatally and in our first years when our stress response and other organ systems are developing and are shaped by what we are exposed to in the environment (stress, infection, toxins etc)
3) the balance between exposure to these kinds of stressors and to “buffers,” such as care from highly connecting, nurturing adults whose low stress nervous systems shape and balance our own capacities for developing resilience to stress as well
A research example on how sensitivity to stress can be shaped very early in life was seen in a landmark epigenetic study out of Mcgill.
http://discovermagazine.com/2013/may/13-grandmas-experiences-leave-epigenetic-mark-on-your-genes
Meaney et al found that mother rats who are more nurturing in the first week of a pup’s life (more licking and grooming behaviors, offering more access for nursing by arching their backs) have rat pups who are less sensitive to stress for life, whereas those who lick and groom and arch back less have pups who are more sensitive to stress for life.
The first study I’ve seen in humans came out in November showing similarities in which mother-baby interactions at 5 weeks of age showed epigenetic changes for genes affecting metabolism and T cell function at 4 years of age (and 3 other genes whose functions aren’t yet known).
http://www.med.ubc.ca/holding-infants-or-not-can-leave-traces-on-their-genes/
Separate bodies of research find that the stress a mother experiences during pregnancy affects birth events (more premature births, complicated births, smaller birth size linked to prenatal stressors etc), her ability to interact with her baby (less nurturing contact, less holding), and that these effects are risk factors for asthma and other diseases. It appears that these are due to altered perceptions of threat (in mothers AND babies) and that repairing the altered perceptions of threat can reverse asthma in kids. If Cortene’s meds are helpful they may be able to help a lot more than just people with ME/CFS.
A book on adverse childhood experiences out last month does a really helpful job of explaining how early events alter the nervous system responses to stress to affect the immune, endocrine and other organ systems over time and helps explain why some of us get sick after stress while others get sick after exposure to toxins or infections etc – and why different diseases can emerge from similar early risk factors.
https://www.amazon.com/Deepest-Well-Long-Term-Childhood-Adversity-ebook/dp/B01N7HZ73B/ref=sr_1_1?ie=UTF8&qid=1518982737&sr=8-1&keywords=deepest+well+book
Wishing this team well in their next steps!!
I would definitely be up for trying this I hate cfs got no life.
For me and my experience, this theory ties everything together. Thank you Cort and Pereira/Cortene. Reading this has given me so much hope.
“also, that under certain circumstances, a corticosteroid (which is essentially a cortisol mimic) could worsen an ongoing stress response and worsen ME/CFS.” I’ve always known this was true for me, I just couldn’t put it together.
I know I should be cautious with my hope, but there are just too many gems in this blog that line up for me. Oh I wish that I could sit at a table with Pereira/Cortene and the Open Medicine-Scientific Advisory Board.
Something to consider: Epigenetic modifications and glucocorticoid sensitivity in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS)
https://bmcmedgenomics.biomedcentral.com/articles/10.1186/s12920-017-0248-3
Thanks, Aaron for bringing this up. I was about to. Corticosteroids and the glucocorticoids Vit. E & A both stir up my chronic shingles on top of worsening other ME/CFS symptoms in me. I was so thankful for the validation of this study, though I’d had to figure out the cause and effect of these drugs and supplements quite painfully on my own over several years. I would need to really understand how they’d test Cortene in the subset of patients who present this way before I’d step up to try it. It’s only my vision and hearing at risk!
Cort’s blog about this ^^^^^^
https://www.healthrising.org/blog/2017/07/30/epigenetics-hpa-axis-chronic-fatigue/
A dim light in the distance, but a light nonetheless; hope is not too late for me. Sounds great, to my uneducated, but very experienced ME brain. This explains why SSRIs made me feel terrible, sicker. I think many of us have been on them, due to the mistaken belief that we are just depressed. I have refused to take them for many years now (my insomnia got too bad), thank goodness. Warning: if you are on SSRIs, don’t suddenly stop them! You should probably be weaned off.
Cort: Just a huge thank you from me. I’ve had some experience in working on my limbic system via neuroplasticity exercises and of all the many many many treatments I have endured, this technique was by far the most effective.
It works on the exact principle of damage to the limbic system.
I think we will see good results. I’m optimistic.
Congratulations Andrea. Can you say what technique you used?
I also get a lot of symptom relief when I practice mindful mediation – body scans especially. After a ten minute sitting, my energy is greatly increased and pem symptoms (that horrible jittery feeling when you are doing too much too) really reduce. If these methods are acting on the limbic system, and we get symptom relief from these methods, it kind of stands to (almost) reason that the limbic system must be a big player in m.e cfs….
It’s called The Dynamic Neural Retraining System led by Annie Hopper. I flew to New Mexico for in person training. It was a hail mary move for me.
In just the 5 days there I had improvement and I learned a lot.
Unfortunately the plan calls for 6 months follow up at home requiring training daily (just 1 hour) along with avoiding anything that could stress out the limbic system. Within a week of returning home my mother was diagnosed with cancer and I spent the next nine months helping her and my father. I was able to because of my gains from the program but was unable to continue the training.
Saying all that, her hypothesis was damage to the limbic system via stress or virus. So I know this is a real answer for us.
Andrea,
I also would like to know which limbic system/neuroplasticity exercises you used. I tried the DNRS (Dynamic Neural Retraining System) system. It worked to some degree and for some time but didn’t completely change things.
its a great article cort and very informative, although i still cant help thinking is the HPA dysfunction driven by an autoimmune condition.
Is our system driven mad by reactions to food, allergy, MCS,…etc
Chicken and Egg really, but i really hope they are right.
Cort, thank you so much for bringing all of these developments to us. This is giving me a lot of food for thought, as it would explain everything for me. This theory also correlates with the series on Vagus Nerve Stimulation you wrote, which I have been using since last year with some benefit I think.
According to the Cortene hypothesis, any calming therapy would be helpful, which Vagus Nerve Stimulation should be, alongside any resurrecting effect on the brain/gut organs connection. I haven’t checked back, are you still getting benefit from VNS?
I need to read the second blog here again to get some of this to sink in. One thing which has occurred to me is the 4-7 day crashes which I experience, where regardless of how my baseline is, I can crash (from usual triggers of activity, stress etc.) to be very ill for around a week. If my baseline is good, then after the crash I suddenly return to something approaching the pre-crash baseline, frequently the 6th morning after the crash. My understanding is that this crash thing is something other people experience too.
Your blog makes me wonder if this period of 5 days is the length of time it takes for the brain to mop up the excess serotonin.
I have reached a stage where I can reach a decent baseline, and see the crashes in isolation. For many years I have had a v low baseline and crashes within the 5 day window, meaning constant & varying illness. And I wonder if the trigger/crash/baseline impact is the pathology of this disease, with many of us in such a fun the pattern is not visible. Unfortunately, to maintain this baseline I have to do nothing, have no stress, no activity, and no life, as whatever is wrong seems still easily triggered. I am working on calming, limbic retraining to try and improve this…
Does anyone else recognise this pattern? Anyone know what makes the quite distinct 5 day length of the crash so distinct? Could it be a serotonin mop up?
Hi Steve,
Yes this is exactly how I experience it too, although for me it seems to be +/- 3 days instead of +/- 5. I’ve had a preliminary theory that it’s the time it takes for the immune system to “run its program”, in the same way as it runs a pretty predictable and similar “program” during a normal cold or flu. But a mop up of serotonin could of course also make just about as much sense. As someone else said, it seems almost a chicken/egg discussion. Both seem involved but not sure which one is the root (if I did you could call me a Nobel prize winner and we’d all pop the champagne! To be honest, which is the root or comes first doesn’t even matter much to me as long as we can find a treatment that works somewhat great. And this theory and coming study, with some caution, seems very promising 🙂
Thanks Secrano, I feel that the length of my crash depends on where my baseline was at, and also the depth of the crash. As I mentioned to Greg below, food can cause deeper ones, and emotional stress too. Curiously heat has caused some of the most surprisingly hard crashes.
I am on something of a mission to see if I can find things that might mop up serotonin, because whilst there doesn’t seem much we could do to hustle the immune system along, which you’re quite right does seem to follow the path of a cold, there are maybe supplements we could take either as a preventative, or when we know its likely we’ve crashed which might help the body mop up serotonin, and whilst this wouldn’t avoid it, it could make the crash shallower and shorter.
As there seems to be some sense to the idea of staying out of a crash can be healing (over 12 months or so), then this might, in theory be a practical tool. I am trying to put a list of supplements together and see if anything has any effect…..
🙂
hi steve,
over weekend i had a trigger food. Today now I have angioedema and relentless fatigue.
It will be thursday before i baseline, gradually creeping up.
To me, this is the length of time the body identifies as “threat over”
Hi Greg,
I feel for you. Food is a mega trigger for me, tho in 95% of cases it is actually from sugar or carbohydrates, which The Cortene theory would explain as an insulin resistance issue… Furthermore, this has eventually resulted in Crohn’s disease as well as IBS type symptoms, which was diagnosed after 10 years, but 6 before CFS, which I think is primary.
Fortunately, removing sugar and processed carbs from my diet can virtually eliminate this type of crash… Tho this is much easier said than done 100%, especially if you aren’t well enough to face cooking properly. It took me 10 years to suss out this elimination was so effective. I have always felt that food was able to be one of the biggest crashers for me of all them, and I think one reason for this is that with activity and stress, these events are more likely to be of a shorter period (not always), and you can kind of remove yourself from them if you are struggling, however sugar/carbs can sit in your gut for 48 hours or more, prolonging exposure to the ‘threat’, and I imagine exacerbating the brain/body reaction.
Have you tried avoiding carbs ever? Or any other elimination diet?
I had to look up angioedema, as I’ve not come across that before, but its difficult when other things add on top of the fatigue. If that is an allergy type symptom maybe some kind of elimination diet would help identify anything to avoid? 🙂
I am trying to avoid crashes at all costs, in the hope that avoiding the hit and run will allow the body to put more distance from the issue and to heal. But avoiding crashes is just so difficult when there are so many triggers, and even with a decent baseline, my crash resistance remains really fragile…
Very interesting indeed. I have taken L-Tryptophan (a precursor to serotonin) for many years to combat depression (works as well as SSRIs with no side effects for me), but does this hypothesis mean that although it helps with depression it could be worsening my M.E.?
That’s what I want to know!
A potentially important link to the quote that cortisol and corticosteroids are mimics is that corticosteroids are known to cause type II (fast twitch) muscle fiber atrophy. A research paper in the 90’s found that 39 of 50 me/cfs patients had type II fiber atrophy. I know this because my recent muscle biopsy revealed severe type II muscle fiber atrophy… it seems possible that increased cortisol could be contributing to this phenomenon and I’m glad to hear that the NIH study is looking at muscle biopsies.
Also Cort I am wondering why you didn’t mention the findings of McGregor that genetic G protein couple receptor mutations were found to be 20 times more likely in ME patients given that from my readings they seem to be so intimately linked to so much of what you talked about in this article. I will have to read again but my take was that the CRFs and some of the others are G protein couple receptors or at least heavily controlled by them…
Thanks for the tip Anthony. I had forgotten about McGregor but I do still remember his enthusiasm about that finding. How nice it would be if his findings and the Cortene work end up meshing…
Does anyone have any thoughts on how (or if)the Cortene hypothesesis relates to the mechanisms of low dose naltrexone? LDN does seem to provide benfits to some ME/CFS sufferers.
LDN is really helping for me. I just started it…(who knew?) My guess – since the HPA axis regulates inflammation then getting it back into control could reduce inflammation – as LDN is believed to do.
I am on LDN have settled on 2mg after 9pm doesn’t help with pain at all but I feel that it has evened out emotional instability and made me more tolerant. I was a screaming wreck if anything went wrong and I found just talking to anyone made me feel extremely irritated Still problems but much improved?
Cort,
Glad to hear that the LDN is working for you. Your hypothesis as to why the LDN and the Cortene could both work on the hypothalmic axis seems quite plausible. I have been on LDN for about 4 months. I was about to give up as it did not appear to be at all effective . In the third month, though, I saw noticeable improvement. Settled on 3 mgs as the appropriate amount, taken at night. I have had post lyme disease treatment syndrome/ CFS for about 22 years. The LDN is the only thing that has provided any relief at all. It is not a total panacea, I still experience epidsodes of profound fatigue but they seem less potent and more intermittent. The Cortene may be an adjunctive thereapy with the LDN that will prove to be the total package for treatment. I hope so.
Great to hear that LDN is helping. Dr. Younger warned about stopping it too soon. Glad you didn’t. 🙂
Hi Cort,
Just following up on your comment last month that the LDN is working for you. My experience is that it generally seems to modulate the fatigue symptoms on an ongoing basis, but very bad days can still flare, without rational explanation. Is that your expereince as well? What dosage are you using? Im doing 3mg, but may go up to the 4.5 level.
Thanks,
Jim
Cogent explanation of the physiology, but how to explain the occasional success of anti-virals? Is it really an equilibrium disequilibrium? I’m skeptical.
Good question. I don’t know…
Perhaps viral reactivation is a stressor that exacerbates ME/CFS, and antivirals reduce reactivations.
How do I become part of this trial?
Details of the small upcoming trial will be provided in the 3rd blog – hopefully in the next week or so.
Just wondering when part 3 will be posted? Did the trial begin? Also wondering why the science media hasn’t picked up on this? It seems to be only being discussed here.
Interested in part 3
When can we hope to see part 3?
Cort–
This is an enlightening summary, I’m looking forward to the next part of this series. Giving what you are highlighting, I’m wondering what attention ME/CFS researchers are giving to an individual’s ability to produce and maintain appropriate levels of BH4.
I wonder if this would help those with dysautonomia (POTS, NMH etc.) since the onset is triggered exactly the same as CFS: post-infectious, physical trauma, pregnancy, emotional trauma etc. Dysautonomia is a dysregulation of the autonomic nervous system which, from what I understand, starts in the limbic part of the brain. I’ll be following this closely!
So this theory turns out to be correct would M.E come under psychiatry or neurology?
Certainly not psychiatry…I would think endocrinology.
Good question!
This study from 1997 in the British Medical Journal points to “Increased brain serotonin function in men with chronic fatigue” This seems rather prescient in the light of this new hypothesis.
http://www.bmj.com/content/315/7101/
Going way back! Thanks Jeremy.
We are all eagerly awaiting part 3 homie.
What has happened to this blog? Will part 3 be available or has something happened to prevent it’s release?
Hi Cort,
I hope you are very well, do you know whether they need venture capital for getting their trials started faster?
Best,
Christoph
Cortene has the money they need to conduct the first trial. If that trial is successful they will need much more money.
I am trying to read this article with my slow brain. But after reading Part 1, I want this drug now! I know that that is reckless and naive that it will work, but I don’t care! I am 39 and have been ill my entire adult life. Getting seriously desperate. I hate having to talk like this. I know others feel the same way.
I totally can relate.
“In fact, depression is caused by high serotonin releases from specific neurons”
That is NOT A FACT. And you are not qualified to say what causes depression.
Could we test the hypothesis regarding serotonin by analysing the effect of taking 5htp supplements (which increases serotonin)?
Afraid not. Cortene believes certain parts of the brain are pumping out too much serotonin (not too little).
This makes so much sense. I have adverse reactions to SSRI’s….it gives me Serotonin Syndrome. I always felt like I had more than enough Serotonin so I have been fearful of trying these drugs again.
Also, after trying Prozac for 7 days and nearly loosing my mind on it…I have never been the same since. Definitely exacerbated my CFS from mild to more severe.
When is part III???
Thank you, Cort!!!!
Sorry. *Losing….not loosing.
1. In addition to the hypothesis that stress/virus/trauma can trigger our chronic state of elevated serotonin, causing CFS,there is also another possible contributor. Overmethylators create abnormally high levels of neurotransmitters, which includes…Serotonin.
2. Is there a lab test that can measure serotonin levels? I know in the article it mentions that there is no “accurate” way to measure this. However, is there a test that gives a “general” idea of what we are workin’ with?
Thank you Cort, excellent explanation of a complicated subject.
It all sounds very hopeful.
Persons with CFS who take CT38 and get relief will still continue to have a genetic predisposition to develop CFS, so that in their lifetime they many need to take CT38 a number of times.
It is not possible to avoid all stressors.
I hope Cortene consider this when setting the price of CT38.
Love the fast-tracking. And the short-term use of the drug.
There is a saying “if the only tool you have is a hammer all problems look like nails”. Well it seems here we have some folks that bought a hammer and looking around started thinking ME/CFS is looking like some kinda nail. HPA axis dysfunction and what not. Now we are getting into speculation that ME/CFS is caused by stress. I am skeptical. I got ME in 1997, it was an intense viral episode that still seems to me spot on with the definition of ME as a type of encephalitis that causes some limited damage to the brain and spinal nerves. I am sure that a lot of us can relate to this experience. I had some profound memory loss including mathematics and corruption of my best talent, which was writing. I am still struggling to write a cogent paragraph. I also lost proprioception in my feet and I had to use google to look it up because my word recall is really spotty.
OK, I am not being negative about this. Cort is doing his usual fantastic job of reporting and explaining something new. I also am eager to read his next installment. And it is very good that some researchers are looking into ME/CFS, cure or not we will learn from their investigation.
I do want to say that I am hearing a lot of people saying things like “CFS has taken away my life” and “This sounds so good I just cant wait for the cure” It makes me sad to hear this sufferring. I know that wishing things were different and hoping for a cure are tempting states of mind. I think it is more effective to try to see things for what they are and then figure out what it is possible to do given that scope and get on with it. Personally if I had spent the last 20 years wishing things were different and hoping for change I think I would have not survived.
I trust this makes a bit of sense and wish you all .he best,
Great series, Cort. And I’m so pleased that after my years of grousing about the ambiguities of the unfortunate term “stress” that you’ve picked up the mantle. Thanks for putting up with me! And then joining the team!
Now how about hat other one, “illness behavior”? : )
Wow this explains A LOT! Especially surrounding the fact that i get SICK every time I’ve gone off Prozac. I was never sick before Prozac, only after it! My poor brain…i wonder how wonderful my life would be right now had i just had the foresight to say NO TO DRUGS as i was taught in DARE.
Now I’m on a low dose of remeron to heal from years or Prozac and to fix my broken sleep cycle. Now this drug can help me heal from remeron use. When does it end?
I’d just like to point out that the only documented cause of reduced 5HT1A receptor sensitivity in humans is SSRI use, and this reduced sensitivity can remain for months after SSRI cessation.
https://link.springer.com/article/10.1007/BF02244438