



VanElzakker’s ME/CFS hypothesis hit a nerve – the biggest one in the body….
In 2013, Michael VanElzakker produced one of the most intriguing hypotheses to date in ME/CFS. His Vagus Nerve Hypothesis proposed that an infection/inflammation near the vagus nerve was causing it to send an unending stream of messages to the brain, telling it to essentially shut the body down by producing fatigue, pain, and other symptoms.
Since then, he’s been particularly interested in the connection between the vagus nerve, the brainstem, and the ME/CFS. He’s not the only one interested in the brainstem.
In 2019 once his brainstem compression was alleviated, Jeff completely recovered from his severe ME/CFS, POTS, and MCAS. Since he published his story over a dozen people have been diagnosed with craniocervical instability – a condition that produces brainstem compression.
In the critical review paper “Neuroinflammation and Cytokines in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): A Critical Review of Research Methods. “VanElzakker et. al. pick apart some of the research done and provide a guide to getting at the brainstem and other regions of the brain they believe have gotten short shrift in this disease.

First, VanElzakker et. al. examines one of the sacred cows in the chronic fatigue syndrome (ME/CFS) community – the preferred term for so many: myalgic encephalomyelitis (ME), which refers to muscle pain (myalgia) related to central nervous system inflammation (encephalomyelitis). While muscle pain is common, it’s not universal. Even people with severe ME may not report pain
Still, the core part of the definition deals with central nervous system inflammation – a description that, with the emergence of the 2015 Yakatomi and the 2019 Younger studies, seems more likely to stick. (A 2018 PET scan study also found neuroinflammation in fibromyalgia as well.)The idea that inflammation plays a key role in this illness makes sense, given the infectious trigger so commonly (but not universally) found.
VanElzakker points to three ways an infectious trigger could produce central nervous system inflammation in ME/CFS:
- Immune factors (e.g. cytokines) triggered by the infection could get transported into the brain. The normally tight blood–brain barrier (BBB) makes blood-borne infections of the brain rare, but the BBB can, like the gut, become leaky in inflammatory states, allowing immune factors and even pathogens entry where they could trigger a large inflammatory response.
- High concentrations of immune factors could allow pathogens to passively diffuse across the BBB,
- Immune factors in the blood could trigger the vagus nerve to send signals to the brainstem and brain, which then sparks an inflammatory response. It’s this last option the authors focus on.
The largest nerve in the body, the vagus nerve transmits sensory, autonomic, immune, and other signals to the brainstem. Even small levels of cytokines in the periphery or body can activate the chemoreceptors in the vagus nerve, which then turn the vagus on – which then activates the immune system in the brain. Studies, in fact, indicate that inflammation in the periphery tends to produce a mirror inflammatory response from the immune cells (the microglia) in the brain.
That brain activation, interestingly, tends to occur in regions (basal ganglia, limbic system organs (amygdala, hippocampus, and hypothalamus), anterior cingulate cortex, prefrontal cortex, and thalamus), that many studies have suggested are also involved in ME/CFS.
The Brainstem
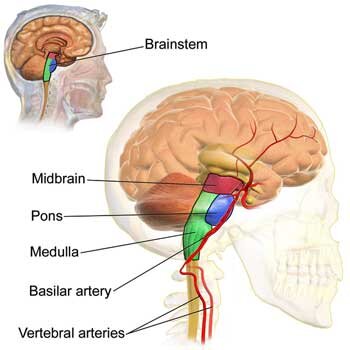 The authors believe the brainstem – found at the base of the brain – right above where the spinal cord terminates – could play a key role in chronic fatigue syndrome (ME/CFS) for four reasons:
The authors believe the brainstem – found at the base of the brain – right above where the spinal cord terminates – could play a key role in chronic fatigue syndrome (ME/CFS) for four reasons:
- Immune signals from the vagus nerve to the brain travel through the brainstem.
- The brainstem is dense with mast cells, and mast cell activation syndrome (MCAS) appears to be common in ME/CFS.
- The brainstem regulates autonomic nervous system functioning – a common trouble area for ME/CFS and related disorders.
- The brainstem also regulates immune functioning; in particular, it triggers an anti-inflammatory response that should limit the inflammatory response.
Seeing the Brainstem in Chronic Fatigue Syndrome (ME/CFS)
Consistent inflammation of the brainstem has not, however, been found in ME/CFS. The authors argue, though, that this is more a case of poor methodology than anything else. The most commonly used method for measuring inflammation in the brain involves measuring the 18kD translocator protein (TSPO) with a PET scan. This protein is produced when the immune cells of the brain – the microglia – become activated. Because the microglia are the chief producers of inflammation in the brain, the TSPO provides an only somewhat reliable way to indirectly measure neuroinflammation and its effects.
The Nakatomi Study
Nakotomi’s small ME/CFS study using TSPO made a big splash in 2014. Tony Komaroff called the finding of central nervous system inflammation the most important study in decades. Five years later, Van Elzakker et. al. called it “important” and potentially “groundbreaking”. The study used TSPO imaging to find widespread neuroinflammation, particularly in the areas leading from the brainstem to the thalamus.
The study was not without significant issues, however. For one, Nakatomi used a spatial “registration” technique that aligns the image on the neocortex or upper part of the brain. This kind of alignment is typically done because researchers tend to focus on the upper, “higher” functioning areas of the brain but is not effective at imaging the lower, more densely packed, primitive areas of the brain, such as the brainstem.
 Nakatomi also used an older tracer (PK-11195) which does not penetrate deeply into the brain and can bind to unintended elements in the brain. Differences in blood-brain barrier permeability between the ME/CFS patients and healthy controls – a distinct possibility – could have confounded the results, as well.
Nakatomi also used an older tracer (PK-11195) which does not penetrate deeply into the brain and can bind to unintended elements in the brain. Differences in blood-brain barrier permeability between the ME/CFS patients and healthy controls – a distinct possibility – could have confounded the results, as well.
Nakatomi’s use of the cerebellum as a kind of baseline measure could have introduced further issues if problems with the cerebellum (another possibility) exist in ME/CFS. Plus, the hypometabolism believed present in ME/CFS could have resulted in lower amounts of the tracer being metabolized than usual – causing higher amounts of the tracer to reach the brain – producing a false positive.
Because exercise may affect how much of the tracer is taken up into the cells, Nakatomi’s use of healthy, non-sedentary controls instead of sedentary controls introduced another issue. Finally, because the brainstem actually pulses with every heartbeat, that movement needs to be accounted for – and usually hasn’t in ME/CFS studies.
The small brainstem nuclei also often not picked up with the standard imaging techniques. Nakatomi’s study results make sense given what we know, and were given a sort of validation by Jarred Younger’s recent results using thermal mapping – a new technology – but better studies are needed.
The takeaway is that the brainstem’s role in autonomic nervous system functioning, immune regulation and the transmission of motor or movement signals – could play a major role in ME/CFS, but is still something of a black box.
Barnden’s Brainstem – the Australian Study
It’s not completely a black box, though. Researchers using other techniques have found evidence of brainstem problems in ME/CFS. Barnden in Australia shifted his MRI to avoid the alignment problem (that VanElzakker mentioned) that prevented him from getting a good image of the brainstem and his studies have found striking brainstem issues in ME/CFS.
One study found that reduced brainstem grey matter volume – suggesting that damage to the neurons in the brainstem had occurred – was correlated with autonomic nervous system problems in ME/CFS. Another which found impaired communication from the brainstem nuclei to other nuclei in the brain suggested the same. Plus it found signs of myelination in the sensorimotor cortex of the brain.
Another which found impaired communication from the brainstem nuclei to other nuclei in the brain suggested the same. Plus it found signs of myelination in the sensorimotor cortex of the brain.
Barnden proposed that decreased signaling from a damaged brainstem provoked a compensatory increase in myelination in the sensorimotor region as it bulked up to try to understand the limited signaling coming from the brainstem. The impaired brainstem-sensorimotor connection might be, Barnden thought, impacting motor functioning in ME; i.e. the ability to carry out physical activity. Signals to move muscles pass from the motor cortex to the sensorimotor cortex down to the thalamus and then through the brainstem to the muscles. (Signals from the muscles to the brain pass up through the same pathways.)
Barnden proposed that the brainstem’s inability to properly relay signals to the motor cortex to activate the muscles could be producing movement problems. His most recent brainstem study validated the idea that the brainstem nuclei and other nuclei in the brain, including the vasomotor region, hypothalamus, and prefrontal cortex were not communicating correctly and were affecting autonomic nervous system functioning in ME/CFS.
Other Kinds of Brain Scans
Other kinds of brain scans, such as magnetic resonance spectroscopy (MRS), can pick up signs of neuroinflammation. Although almost 10 MRS studies of the brain in ME/CFS have been done, VanElzakker et. al. report that a clear and consistent picture of metabolite alterations in the brain has yet to emerge. They believe that’s due largely to a common theme in medical research – lack of standardization. Different diagnostic criteria, different types of healthy controls, different brain regions examined, and different metabolites targeted make it difficult to present a clear picture of the metabolic alterations in the brains of people with ME/CFS.
The Japanese Take
The Japanese probably couldn’t agree with Barnden more. Their studies indicate that, as the healthy controls became more fatigued, two core regions – both of which communicate with the brainstem ( the prefrontal cortex and the anterior cingulate cortex) – shut down.
As these regions begin to shut down, control of autonomic functioning becomes lost. In particular, the ability to activate the parasympathetic nervous system (i.e. the vagus nerve) and tone down the sympathetic nervous system activity, disappears.
They propose that a breakdown in what they call the facilitation system has occurred. As we become fatigued, the facilitation system increases the signals going from the primary motor cortex in the brain to the muscles. This increased “drive” tells the muscles to work harder and activates more muscle cells.
So long as new, fresh muscle fibers can be recruited, the exercise can continue. If no muscle fibers are left to be recruited or if the brain has a problem recruiting new muscle fibers, fatigue sets in.
A 2003 study “Deficit in motor performance correlates with changed corticospinal excitability in patients with chronic fatigue syndrome“ suggested that reduced muscle recruitment caused by inadequate motor cortex inputs was indeed occurring in ME/CFS. That authors proposed that “changing motor deficits in CFS has a neurophysiological basis [which] … supports the notion of a deficit in motor preparatory areas of the brain”. That study,was apparently never followed up on.
Conclusion
Several studies suggest significant brainstem issues may be present in ME/CFS and could produce everything from autonomic nervous system to immune system issues to problems with movement.
In this review article, the authors critique past brain imaging studies and provide a “how to” guide to assess the brainstem in ME/CFS. Barnden’s Australian brainstem studies suggest that when done correctly, MRI imaging studies are finding brainstem damage and have produced evidence of brainstem neuron demyelination, a compensatory remyelination in parts of the brain the brainstem connects with, and reduced connectivity between these regions.
The authors propose that imaging studies focused on the specific pathways in the brain – two of which include the brainstem – that have become activated by inflammatory processes should be able to pick up the neuroinflammation they believe is occurring in ME/CFS. Lastly, the authors suggest targeting where the vagus nerve enters the brainstem (the nucleus of the solitary tract (NTS).



I have M.E/CFS so please let me know if I can join a clinical trial. I’m in London
Can you recommend any treatments to try for my CFS/ME and pain brought on by an Epstein Barr virus, 20 years ago. 10 years ago, the EBS reactivated in my body. The fatigue and lack of energy is s bad, functioning is a huge challenge. I’m single and live alone. Any suggestions?