


Sustained Stimulation of β2- and β3-adrenergic Receptors Leads to Persistent Functional Pain and Neuroinflammation. Xin Zhang, MD, PhD, Jane E. Hartung PhD, […], and Andrea G. Nackley PhD, PhD. Brain Behav Immun 2018 Oct;73:520-532. doi: 10.1016/j.bbi.2018.06.017. Epub 2018 Jun 20.
There’s neuroinflammation and there’s the stress response. Recently, we saw Mackay and Tate propose that neuroinflammation in ME/CFS and FM was linked to a whacky stress response centered in the hypothalamus.
In 2018 Chinese-Duke University collaboration suggested that the stress response and neuroinflammation are linked as well, although in a different way. The appearance of these hypotheses and findings is good news given that neuroinflammation appears (but has not been definitively proven) to be present in these diseases, and evidence suggests that the stress response is certainly off.
Studies suggest that the activity of the “fight or flight” (sympathetic nervous system) response is increased and/or that the activity of the “rest and digest” (parasympathetic nervous system) response is decreased in ME/CFS and FM.

This Chinese-Duke collaboration proposed that an overactive stress response system (“enhanced catecholamine tone”) has triggered the neuroinflammation in fibromyalgia (FM), chronic fatigue syndrome (ME/CFS) and similar disorders. No injury was necessary to start the just a really amped up fight/flight system.
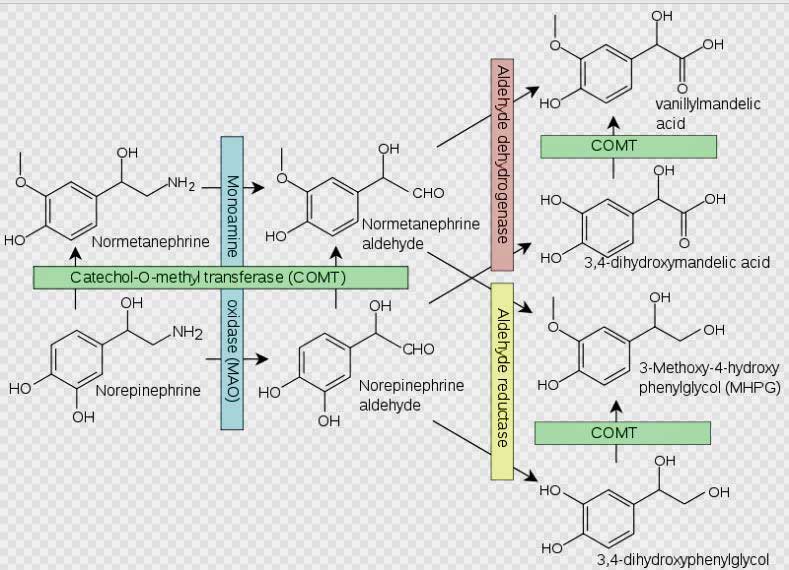
Norepinephrine breakdown by the COMT enzyme. Problems with norepinephrine breakdown by the COMT enzyme are suspected in FM and ME/CFS.
The inability to find an immediate physical origin for the pain in FM and other “functional pain” diseases (temporomandibular joint syndrome (TMJ), vulvodynia, irritable bowel syndrome, interstitial cystitis, chronic fatigue syndrome (ME/CFS)) has thrown the medical profession for a loop. Too often, that has led doctors to dismiss the pain and other symptoms in these disorders as psychological or to treat it as something not significant.
Over time, however, consistent problems in the HPA axis, the sympathetic nervous system (SNS) and central nervous system have been found which could help explain the pain. While it’s possible that the body pain in FM is to some extent being generated in the body, it’s clear that it is also being generated in the brain;; i.e. pain is being brought to the body by the brain.
The Gist
- Many studies indicate that prolonged activation of the sympathetic nervous system (SNS) can result in increased pain sensitivity
- The researchers in this study amped up the SNS system of rodents by inhibiting the activity of an enzyme called COMT that breaks down norepinephrine – the main driver of SNS activity.
- It took from 2-3 weeks for a chronic pain state and signs of neuroinflammation to appear – a state which remained even after the researchers turned off the sympathetic nervous system activation.
- That indicated that while the SNS activation was needed to trigger the chronic pain state, something else, after a time, maintained it in the brain.
- The authors found that three cytokines (TNFα, IL-1β, and IL-6) were driving the chronic pain state in the brain.
- They were able to stop the pain by inserting a TNF-a blocker called etanercept (Enbrel) into the rodents spinal cords.
- That suggested that while the SNS activation triggered the pain state, attacking the neuroinflammation may be the key.
- Enbrel is used to combat pain in several autoimmune diseases but does not work in everyone and can have significant side effects.
- The authors proposed that drugs which target specific parts of the TNF-a immune pathway would work better.
- Dr. Nancy Klimas’s modeling efforts suggest something similar is happening in ME/CFS and FM. She’s using Enbrel plus an HPA axis drug to tamp down the neuroinflammation first and then hopefully reset the HPA axis in GWI and ME/CFS.
Well over a dozen studies have explored whether a COMT polymorphism – an unusual form of the gene – could be playing a role in fibromyalgia. Most studies have found links to COMT polymorphisms in FM, but not all. (It was remarkable to see two meta-analyses done a year apart come to opposite conclusions concerning whether the val(158)met COMT polymorphism plays a role in FM.)
The geneticists are not giving up, though. One of the latest studies, which examined almost 3,000 people with FM, gave a nudge to the COMT/FM hypothesis and concluded that, yes, more studies were necessary.
Possible problems with both COMT and the adrenergic receptors have shown up in ME/CFS, which also appears to share the fight or flight system activation found in FM. Wyller found an increased incidence of unusual forms of the B2AdR and COMT genes (polymorphisms) in adolescents with ME/CFS in 2011. The next year, the Lights found that exercise dramatically increased the expression of beta adrenergic and other genes in ME/CFS patients, but not in healthy controls. Just recently Loebel and Scheibenbogen found increased antibodies to B2 receptors in a subset of ME/CFS patients.
Both Loebel and Scheibenbogen and Wyller’s findings suggest that the SNS problems don’t stop at that system and that an SNS/immune connection is present in ME/CFS. These Duke/Chinese researchers would agree.
Sympathetic Nervous System – Neuroinflammation Connection Found
“In line with findings from studies of inflammatory and neuropathic pain (Latremoliere and Woolf, 2009), our results suggest that functional pain is maintained in the central nervous system long after the precipitating cause is removed. This phenomenon may explain why patients with FPS (fibomyalgia pain syndrome) experience sensory abnormalities at regions remote from the original painful site.” The authors
The Duke/Chinese researchers were intent on determining how sympathetic nervous system activation could lead to immune activation, long-term neuroinflammation and chronic pain.
They turned up the dial on the sympathetic nervous system by inhibiting the COMT enzyme that breaks down the norepinephrine and then they waited and watched. Fourteen days later, their subjects – rodents – were demonstrating signs of widespread pain.
This was not a surprise. Numerous studies have linked the activation of the adrenergic B2 and B3 stress receptors that drive the fight or flight system with increased pain sensitivity. These receptors appear to be producing pain by increasing the excitability of pain signaling neurons and by activating T-cells, mast cells and others. (Interestingly, this study found that activated B2 and B3 receptors in the body – not in spine or supraspinal regions – triggered the increased pain sensitivity.)
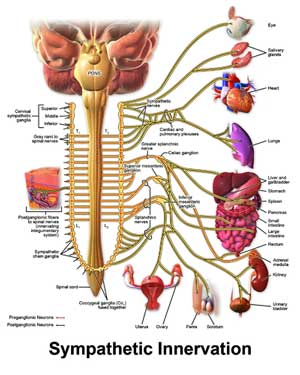
Mostly we think of the sympathetic nervous system’s effect on the body, but these researchers found that chronic SNS activity in the body could affect the brain as well.
Importantly, they showed that constant sympathetic nervous system activation induced a chronic pain state that had a life of its own. Even after the COMT inhibitor was removed the rodents’ pain levels remained high.
Next, they tried to determine what was driving the chronic pain state. Turning off the B2 and B3 AR activation with antagonists didn’t do anything to the pain. That indicated that while the adrenergic receptor activation played a critical role in triggering the development of the pain state, and had apparently strengthened the “pain-coding pathways”, something else was now driving the chronic pain state in the brain.
That something else proved to be three cytokines: TNFα, IL-1β, and IL-6. As the brakes were taken off the COMT enzyme the cytokine levels in the plasma decreased but 1-3 weeks later continued to increase in the cerebral spinal fluid. About the same time, the immune cells in the brain called microglia and astrocytes showed signs that they were activated.
The researchers had just shown how to induce a state of neuroinflammation and chronic pain without ever causing a physical injury. Note that the neuroinflammation in the rodents’ brains was simply initiated by increasing the levels of epinephrine and norepinephrine (by inhibiting the COMT enzyme).
Tricky Situation?
Wyller showed how much of a difference a slightly altered gene can make – and just how tricky this situation may be in ME/CFS. He found that the activity levels of ME/CFS adolescents with a high activity (Rs4680) form of the COMT gene tanked when given Clonidine.
That was a fascinating result given that Wyller believed the opposite would happen. With his data indicating that high norepinephrine levels were present in ME/CFS, Wyller believed that Clonidine would decrease the adolescents’ sympathetic nervous system activity, increase their parasympathetic nervous system activity – and provide them with more energy. Clonidine did decrease the SNS activity of this particular group of patients, but to such low levels that they actually got worse. It turned out that this group already had lower NE levels.
Wyller’s finding in two groups of ME/CFS patients, one with higher and one with lower NE levels, and each with different immune factors at work, suggests that the situation may be quite complex.
Treatment Possibilities
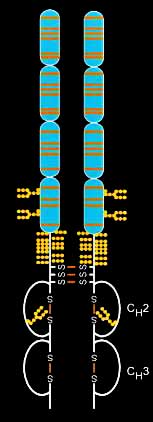
Enbrel – injected into the spinal cord – was able to turn off the pain state in the rodents.
The researcher were able to stop the pain by administering either of two substances – the TNFα inhibitor etanercept (Enbrel) or the p38 inhibitor SB203580. That indicated that TNFα and p-p38 were driving the chronic pain state in the brain.
Enbrel is used in the treatment of inflammatory pain conditions, such as arthritis and inflammatory bowel disease, and neuropathic pain conditions, such as lumbar disk hernia and sciatica. About 40% of the patients, however, do not respond to TNFα antibodies and long-term use is associated with an increased risk of infection and other adverse events.
The authors suggested that drugs which selectively target parts of the intracellular TNFα signaling network could produce better results and fewer side-effects for people in chronic pain.
Dr. Klimas’s models, however, suggests that using etanercept (Enbrel) first and then using the HPA axis modulator next, Mifepristone, could flip the systems of Gulf War Illness and ME/CFS patients back to normal. She is testing that model now.
Conclusions
The sympathetic nervous system route to chronic pain is being increasingly well documented, with the authors able to provide many citations to support it.
It should be noted, though, that this was a rodent study, not a human study. Except for Enbrel, the authors were unable to point to TNFα altering drugs that might work. The authors’ ability to insert Etanercept straight into the brain obviously also couldn’t be repeated in humans.
Nor have the genetic issues with the COMT enzyme or the functioning of the beta adrenergic receptors been completely fleshed out in FM or ME/CFS. Some studies suggest the situation may be complicated by different subsets of patients.
The authors also don’t tell us why the stress response might get lodged on the “on” position long enough to get an immune response in the brain going, nor if people with FM are more susceptible to getting neuroinflammation started in the first place.
It’s interesting, though, that two things that came out of the Dubbo post-viral studies – that symptom severity during an initial infection, plus the presence of polymorphisms that could have pumped up cytokine production – suggested that an early over-active immune response to the infection could have paved the way for ME/CFS . One wonders if something similar will be found in people having trouble recovering from COVID-19.
- On that note check out a really excellent article “COVID-19 Can Last for Several Months”. It’s the fourth one in the last month or so to focus on difficulty some people are having recovering from COVID-19 and it explicitly mentions ME/CFS.
FIrst and foremost the authors believe that brain inflammation in functional pain states like FM and ME/CFS must be addressed
Still, the study presents a pretty compelling picture of how an upregulated fight or flight system could lead to a chronic pain state maintained in the brain by immune activity. It suggests that pain state may be triggered by receptor activation in the body, not the central nervous system, and that a successful treatment needs to target both the neuroinflammation in the brain and the SNS activation in the body.
Dr. Klimas’s modeling work has come to a similar conclusion. She’s attempting to tamp down the neuroinflammation in the brain using etanercept (Enbrel) and then reset the HPA axis in Gulf War Illness and ME/CFS.



So much fascinating research continuing to emerge. The question about how the stress response might get lodged in the “on” position is right up Dr. Robert Naviaux’ alley with the cell danger response. It is also up there with what we’re learning from the science of adversity, such as how early life stressors (prenatal, birth, first years of life, childhood) can “program” the HPA axis to work in certain directions, including heightened sensitivity to stress and a more survival-oriented stress response. We’re also finding that gene function needs interactions with environmental factors to manifest so this may be among the many complex factors influencing our health and ME/CFS.
This dratted stress response is so hard to turn off that it feels like some hardcore programming has been inserted. God knows for what reason. For some the changes in programming began early on and then apparently went ballistic when the right situation presented itself. For others it happened later. I sure hope COVID-19 gets the medical community looking in our direction.
I wish it was not complex but it probably will involve a variety of factors. As a twin I wonder what factors came into play for me which didn’t involve my brother.
Yes – these questions of early events setting up programming are pretty interesting. along with the Why – Naviaux and others talk about it as programming for the environment we’re being “trained” due to stress to expect.
I’ve created a new construct called Adverse Babyhood Experiences (ABEs) for events up until our 3rd birthday that builds on the research showing how early adversity shapes health.
Some of the twin studies have found that it’s the smaller twin, or the one who might have needed more care or longer care (such as intensive care r incubator care) or who was the sickest that may have experienced more “hits” and therefore be more vulnerable. It could also be if one twin was less “bonded” or had a parent that attached less to them than the other (it’s often hard to attach to more than one baby at a time).
No idea what your experience was…but I imagine it was complex in its own way!
Thanks for this Cort. Really go article as always. So good to hear research is getting nearer to understanding some possible causal pathways. Does this link to the Cortene model in an earlier blog? Any updates on their clinical trial?
@Alison:
“Does this link to the Cortene model in an earlier blog?”
I remembered the Cortene hypothesis being related to serotonin going wrong. I also vaguely remembered from recent research that serotonin and COMT are somehow related.
When looking COMT up, there was no mention of serotonin. Rather then giving this idea up I delved deeper.
Looking for serotonin and COMT yielded nothing, but spelling COMT full did yield a paper titled
“Serotonin-Induced Hypersensitivity via Inhibition of Catechol O-Methyltransferase Activity”
With in it
“Here we show that serotonin also inhibits catechol O-methyltransferase (COMT), an enzyme that contributes to modultion the perception of pain, via non-competitive binding to the site bound by catechol substrates with a binding affinity comparable to the binding affinity of catechol itself (Ki = 44 μM).”
Says plenty I think.
with first author “Douglas Tsao”. I don’t link the NCBI paper as that’ll get my comment blocked again for moderation. It should be easy enough to track the paper however this way.
Now I was left to check if Cortene’s hypotheses involved too few or too much serotonin. From https://www.healthrising.org/blog/2018/02/17/cortene-chronic-fatigue-syndrome-hypothesis/ it seems it involves too much serotonin.
(and that would lead to stronger COMT inhibition according to the NCBI research paper).
@Cort, please correct me if I am wrong on this “too much” as I’m too close too crashing to spent much effort into looking deeper.
If so, The Cortene hypothesis may involve and or be a side track of the lengthy DOPAL / acetaldehyde / adrenaline / COMT vicious circle described by me below.
The several parts below may link several previous separate ME hypothesizes for those with the stomach to wade through it. All have fairly substantial research paper backing if I didn’t screw a thing up. I left those references out as writing that down borderlines the maximum I can do in a single go without crashing (on a combined dopamine adrenaline high ;-)).
The fifty plus references also would get it blocked for sure and make it unreadable. I plan to move part of it with proper references later to the general part of the forum.
Interesting to hear of others experiencing that internal tremor. It can be quite severe when the body has been overtaxed or stressed. An awful sensation….there is no way you can relax or sleep. The only thing that will help at all is Valerian.
I remember (anyone else recognize what a great, if rare, statement that is? Lol) seeing a 60-Minutes video out of Australia several years ago. It was about a doc in FL who was injecting Enbrel into the CFS (cerebrospinal fluid) of stroke patients and putting them in an inverted position to get it more directly into the brain. He had some rather dramatic positive results, at least as shown on the program. May not be totally out of the realm of possibility to get it into the brain in humans, even if it can’t be directly injected into the brain.
Thanks for yet another great article linking various studies that all seem to hold some promise for future treatment ideas.
Whoa! That guy had the courage of his convictions. That’s really something!
Wondering if the lower Cortisol levels and higher RT3 (reverse T3) levels, both often found in ME/CFS patients, contributes to the perseverance of the increased epinephrine levels. HPA axis: hypothalamus pituitary, adrenals? High RT3 also a signal of stress.
Sorry but I had ME 28 years ago and I believed it to be a disease. Now i think it is just hypothyroidism with a conversion problem T4 to T3 https://www.youtube.com/watch?v=CVXvYrJJ5dU&t=2154s
https://www.meassociation.org.uk/2018/03/frontiers-press-release-chronic-fatigue-syndrome-possibly-explained-by-lower-levels-of-key-thyroid-hormones-20-march-2018/
There are some very ill people who need T3 replacement therapy urgently.
I agree with almost everything you said and thanks for passing this on. We did a couple of blogs on this:
https://www.healthrising.org/blog/2019/03/04/hypothyroid-chronic-fatigue-syndrome-thyroid-ntis/
Martynas has really dug into it:
https://www.healthrising.org/blog/2019/03/07/thyroid-t3-chronic-fatigue-fibromyalgia-recovery-stories/
I don’t think it’s happening in everyone but it’s certainly something that everyone should check out.
Well it might be a disease What’s happened with me is I recovered for the last 13 years Now it’s come back in a nocturnal form. I feel pretty much ok during the day but at night during sleeping I wake up feeling like a tremor through my body as if electric is attached. I don’t physically shake like Parkinson’s but it feels like I am inside it’s been freaking me out for two weeks. Also kind of feel a bit itchy sometimes or tickley perhaps. Sometimes I have startled or jerked awake. I went to the hospital and they said it’s restless legs syndrome although it’s not really my legs. This is what made me think about low T3 Would low T3 cause nocturnal symptoms? I’m in the UK and no GP believes in ME CFS. They pretend they do or know it but they’ve been led to believe it’s psychological and harmless. Has anyone had these symptoms. Thanks.
@Martin, yes I have those tremors too. And they are internal feeling, though at times mine are external too. For me, it is mast cell connected. And there is no sleeping with them. And there can be a bit of a falling, jolt awakening. It is most miserable. I have them awake also. But I’m getting close to having that figured out too and under more control.
Histamine can also help to clean up acetaldehyde. So there is a connection there.
Well, I’ve been taking supplemental T3 for years. I took so much of it that I overdosed on it, and had to re-balance with more T4 and less T3. And I still have ME/CFS.
To add to my post above, I’m no doctor but doesn’t the adrenals kick in once you wake and get up? Mind you my TSH and adrenals were normal in the past. But I’m wondering if that accounts for why I feel ok during the day and feel tremor at night Strange how it seems to fit.
@Martin, most of our detox happens at night. We do it while sleeping and if not sleeping……we seem very aware of it. Especially us super sensitive ones.
I agree that T3 conversion problems can exacerbate ME, & that in some cases, the problem probably is entirely conversion issues. However, I have dealt with such conversion issues for many years now, & am on T3 & my levels of TSH, free T4, Free T3, reverse T3, etc are all very good, & I am most definitely still very ill with ME.
I agree as well . The fact that the HP axis is not working properly is shutting down our endo system. Now we are left with thyroid and adrenal insufficiency . The problem with cortisol is we do not know how to supplement it properly . If there was better technology allowing us to supplement cortisol in a pulsatile state on demand when our body needs it this disease would be a thing of the past. The proper homeostasis of cortisol is crucial for survival without it we are up s—- creek with out a paddle. If you look at people with actual dx adrenal diseases they are having the exact same symptoms as people with ME. What people with adrenal diseases are doing now including myself are using an insulin pump to pump liquid cortisol . With the pump you can create a cortisol day curve with the basal rates . This method of treatment is drastically improving peoples lives . The NIH has done a few studies on this method and have concluded that the pump lowers the risk of adrenal crisis bye up to 75% and lessons general side effects of the steroids up to 60%. Again its not the drug its self that is the problem cortisol has to be supplemented the way nature intended and that is pulsatile . Researchers know now that the circadian rythum of cortisol is crucial for survival. The Mayo clinic has reported that researchers are working on a nano pump that will be implemented under the skin and release cortisol in a more specific manner closer to our rythums. Technologies specific to drug delivery has huge potential for hormonal diseases . Advocates like Paul Robinson for example have been fighting for this for decades now. We absolutetly can get way better with a different approach to hormone supplementation. ME is the only disease that is large in part due to adrenal insufficiency and does not recieve treatment thats insane how can people in this century 70 years after the discovery of cortical steroids still be dieng from adrdnal disease . Before the discovery of steroids a persons life expectancy was 3 years with adrenal disease . To see theses severe ME patients left for dead is heart breaking and in my opinion its malpractice .
When are Klimas’s results due? This trial seems to have been talked about for years.
Great question. It has been quite a while. I believe she has some results back from the GWI trial but I don’t know how close we are to finding out what happened or what’s going on with the ME/CFS trial. Everything always seems to take longer than expected.
Hey Cort/Zen,
I joined the INIM webinar Dr. Klimas’s team held three weeks ago. I asked if there were any updates on the etanercept & mifepristone ME/CFS trials that have been reported to be underway this year at Nova on the webinar.
This was Dr. Klimas’s response to me, if this helps shed any light on the situation:
“We had a delay in start up due to some IRB (human safety review ) delays then the COVID-19 epidemic, we were just about to start the first subjects the week that everything shut down. We anticipate a restart in the month of July we will be recruiting men and postmenopausal women into this open label study. This is a pilot funded by private donations, we are also applying to the NIH next week for sufficient funding to move the project along and expand to the other subgroups”
Hope this helps for an update.
Thanks Dakota! I hope she can get that funding. Perhaps the NIH will view ME/CFS as more of a priority with the COVID-19 recovery problems.
Is the duo of drugs in trial by Dr. Klimas working? Has anyone tried them “off-label” and experiences any benefit – do we know?
Hi all, this is really interesting. I am just wondering how this connects with the use of Cymbalta, an SNRI, to treat FM? Wouldn’t the use of an SNRI actually increase norepinephrine signaling, which should make things worse based on these study results? Maybe I am missing something 🙂
Did you get a reply to that question?
Yes, SNRI’s should increase both serotonin and norepinephrine levels. I have been repeatedly told, though, that no one actually knows how they work. The upregulation of the animals SNS was triggered in the body not the brain. I’m not sure how they fit or don’t fit with this. They can certainly help some people with FM.
I have very slow COMT and when I took Cymbalta I felt horrendous. It was a traumatic experience which I endured for months upon guidance from my doctor. When I got my genetic testing years later and discovered slow COMT, my new doctor suggested that’s why I reacted so bad. WAY too much norepinephrine in an already sympathetically exhausted system.
Cort, any mention on beta blockers for high blood pressure exacerbating the fight flight feeling? Sincerely, Javen I was taking propanalol specifically for the fight flight feeling but it’s still there. Sincerely, javen
Because propanolol does inhibit the effects of norepinephrine one could hope that it would calm things down. I think in some people it does. It’s quite a complicated system, though.
And the additional NE could be a compensation. It helps vasodilate and increase blood flow. Which would in turn help carry oxygen to much desired places, like heart and brain. Though very uncomfortable.
I have had ME/CFS for about 25 years and from the start, have had a great reduction in fight or flight capacity, as well as becoming hypo-thyroid and hypo in terms of adrenal function, as well as with other hormones. Finally last year had epinephrine and norepinephrine and dopamine levels tested: first two are “clinically undetectable” and last (dopamine was very low). Everything about me is low and slow.
While I can see how people with FM have and activated SNS, I wonder about the data for people with ME/CFS. For instance, how many patients were tested and how long had they been sick?
I also have quite a lot of pain, including small fiber neuropathy. For instance I can only tolerate a very few kinds of fabrics and clothing.
I can’t discount that in the run up to my illness, I may have had an activation of the SNS which led to those changes, but what about since then when my fight or flight has been so tanked?
I tend to think that in the desire to connect a lot of dots in understanding these illnesses, as well as to find successful treatments, that conclusions can be rushed and assumptions made. For instance, if in FM there is SNS activation plus pain, then could some researchers be hypothesizing that what holds there may also be true for ME/CFS because there is also pain in ME/CFS? But I am sure there are many like me with ME/CFS who don’t have a vigorous adrenal system and high levels of those hormones, or maybe not since early in their course or prior to its beginning.
Of course I haven’t seen the details of all the human studies you mentioned and probably wouldn’t understand them really well if I did. I did feel it worth saying, however, that not everyone with ME/CFS or who knows of patients with it would see most, and certainly not all of us as having a high level of SNS activation.
You are very hypo! I’m surprised that more studies haven’t examined NE and E in ME/CFS. I don’t know that we have a lot of data on that. Wyller did find a lower NE group in ME/CFS which had different immune issues than the higher NE group.
The heart rate variability studies are where the over-active SNS / underactive PNS really shows up. I don’t know if you need high NE levels to have low HRV or not.
Thank you for writing about this complex topic,
I think it is important to use stringent diagnostic criteria in ME-research.
That appears not to be the case with the two Wyller-studies referred to:
In the study «Genetic variation in catechol-O-methyltransferase modifies effects of clonidine treatment in chronic fatigue syndrome” published in 2016, 120 CFS patients aged 12–18 years with 3 months of unexplained disabling, chronic/relapsing fatigue of new onset were included. No other accompanying symptoms were required.
“Polymorphisms of Adrenergic Cardiovascular Control Genes Are Associated With Adolescent Chronic Fatigue Syndrome” published in 2011, required 4 months of chronic or relapsing fatigue and no accompanying symptoms.
Yes, Wyller was way out there on the skinny branches for quite awhile – using his own definition of ME/CFS. Since that meant his results couldn’t really be compared to the vast majority of other ME/CFS studies, I can’t fathom why he decided to undercut his work that way. I think he is using more accepted definitions now.
Unfortunately, he is not. Wyller has set the premises for the approach, and treatment of ME patients in Norway for 20 years. His research is not even relevant to the disease.
I have gone through 39 of his articles and found that none of the studies required more than a few months unexplained fatigue with no accompanying symptoms. Read more here:
https://melivet.com/2020/06/03/me-expert-is-not-researching-me/
I have also analyzed his latest article where adolescents were treated with music therapy, and it is not very impressive to say the least:
Link to English translation included:
https://melivet.com/2020/05/08/kurere-me-med-musikk/
My mistake – For some reason I thought he was using standard criteria but apparently he’s not. I can’t for the life of me understand why he doesn’t just use standard criteria. I haven’t seen anyone else just make up their own criteria. Perhaps the mark of a stubborn personality?
THe study is all very well and good and seems right on target now except one thing. I never got pain when I had ME/CFS. Well I tell a lie. I used to sometimes feel neuropathic pain in my legs which came and went but pain was never a big factor in my ME/CFS. My main problem was feeling lightheaded standing up and cognitive problems as well as the fatigues. These researchers IMO might be researching FM, possibly GWI as stated rather than ME/CFS but I don’t know.
The other thing is my thyroid chemistry was always normal as should most people’
s with a diagnosis of ME/CFS (you can’t technically have acute onset ME/CFS if your thyroid is underactive by definition.) So how is it that some people report HPA axis dysfunction? I used to feel chills when I was in stage one of ME/CFS sometimes but still my thyroid TSH was normal. ANd I believe my adrenals were normal too (sorry 28 years ago now so difficult to remember).
It is now my view (changed somewhat over the last 24hrs lol) that people taking T4 and T3 do not need it if they have ME/CFS contrary to Dr Skinner’s theory that ME/CFS is just a form of hypothyroidism with a normal TSH. WHy so many people (mostly in America I think) are taking thyroid meds I can’t understand. Once again, for ME/CFS acute onset, your thyroid must be normal.
Now as stated above, I have a theory for my current nocturnal symptoms many years after recovering from ME/CFS and I said is it the adrenergic system/hormonal system malfunctioning at night causing only nocturnal symptoms? I can’t understand it!.
Pain is actually one of the most common symptoms in ME/CFS but not everyone has it. It is one of my biggest symptoms.
I think it’s quite possible to have mostly nighttime issues. Some people have really dramatic night-time sleep problems – weird wakenings, rapid heart rates – stuff like that.
”Studies suggest that the activity of the “fight or flight” (sympathetic nervous system) response is increased and/or that the activity of the “rest and digest” (parasympathetic nervous system) response is decreased in ME/CFS and FM.” This is what i believe too.
But why? Could it be a compensation? Or the high fever most patiënts have had. Anyway this system is broken. All other problems in our body is due to this mechanisme. We’re in a loop 🙂 We can’t come out.
In one of his studies Wyller said he believed that the SNS upregulation was compensatory: i.e. the SNS is not broken – it’s trying to stabilize the body in some way. That’s seems like a totally valid possibility to me.
May I refer to the recent long list of comments Issie and I made on
https://www.healthrising.org/blog/2020/05/27/neuroinflammatory-paradigm-chronic-fatigue-me-cfs/
We had little response to it but these comments actually talked about our recent findings where the COMT enzyme and genes AND how that could be caught into a vicious circle were at the core.
I’ll leave the technical links to a follow up as I want this comment to pass ASAP without moderation and those links get me often blocked.
The COMT genes are not only important for (nor-)epinephrine metabolism but also for dopamine metabolism. Actually nor-epinephrine also better known as nor-adrenaline is directly made from dopamine. And the COMT genes and enzymes play a direct role in degrading and detoxing dopamine.
There are several routes in degrading dopamine. The three main ones are:
A) By the enzyme dopamine beta-monooxygenase directly making nor-epinephrine from dopamine.
B) By a chain of three enzymes of which the first one in the series is COMT creating fairly benign 3-methoxytyramine from dopamine
C) By a chain of the same three enzymes as in B) of which the first one is MAO creating DOPAL, a *VERY* toxic chemical suspected to be strongly involved in Parkinson Disease.
Now if COMT for some reason is “slower” then route B) will go slower. That will “push” more dopamine in the remaining pathways A) and C). In plain speak: that will create more nor-epinephrine and more *VERY* toxic DOPAL.
The current blog discusses increased nor-epinephrine. That directly links to these ideas.
Increased DOPAL directly links to our ideas. DOPAL needs COMT and ALDH for further degradation. In our ideas we also hypothesize that a strong flare of poor to very poor local oxygenation is part of the recurrent crashes and PEM we ME patients experience. Some of us experience extreme air hunger during the worst crashes even “if we breath our lungs out”. But there is far more to this idea then just this observation.
The problem with ALDH is that it requires oxygen (inderectly in the form of NAD+) to work. ALDH requires NAD+ that is in very short supply when our bodies go deeply into anaerobic metabolism / glyoclysis. That is bound to happen far more in us during the combination of poor oxygenation with exertion. Those conditions are very close to strong crash conditions and go hand in hand with lactate buildup (in the brain).
When somehow we are already prone to poor COMT functioning then we already produce more DOPAL to start with as less dopamine will be degraded by the more benign 3-methoxytyramine pathway AND the DOPAL detoxification will be slowed by less COMT activity (3th detox step) having a double effect on increased DOPAL baseline levels.
When we, our brain and / or in more extreme conditions our liver get into deep anaerobic metabolism then this DOPAL detoxification to far less harmful chemicals is slowed down a lot as the next (2nd) detoxification step does require oxygen in the indirect form of enough NAD+ which is in short supply during deep anaerobic metabolism.
DOPAL is not only extremely toxic, it is also known to be able to:
* destroy motor (and dopaminergic) neurons when building up too high (like in Parkinson Disease)
* to strongly inhibit mitochondria
As dopamine is higher in “dopaminergic neurons”, including very strongly the motor neurons but also the hypothalamus and other key brain parts, it is reasonable to assume that dopamine degradation products concentration like DOPAL will be highest in these neurons during and after a strong crash.
This could help explain the very very weak limb strength and often even short lasting leg paralysis I experienced after a crash, just as well as these key areas in the brain like the PVN as Angus Mackay suggested among other key “primitive” brain areas.
Why would it affect all of those the worst? Because too high DOPAL has a strong to very strong inhibiting effect on mitochondria.
Note that that may be a “life saving” mechanism as in our ideas this selective mitochondria inhibition prevents further and chronic buildup of excessive DOPAL, likely preventing much of the permanent damage by neuronal death as seen in Parkinson Disease.
We also have identified a mechanism where too high DOPAL creates more DOPAL creating not only a vicious circle but slowing building this too high DOPAL concentration down leading to prolonged limb and mind weakness as is seen in PEM.
We have also documented pathways that could help explain why this too high DOPAL concentration acts only in a delayed fashion into full force, giving us often – not always delayed as I personally experienced sometimes – delayed PEM onset.
Hi Dejurgen.
Great ideas. These are difficult concepts so might explain the lack of feedback.
Further to your comments, I think there might be a tenuous link between high sensitivity / giftedness and ME/CFS and I also sometimes wonder if COMT / MAO polymorphisms that slow down NE and dopamine breakdown may play a role in the illness.
You didn’t mention in this particular comment but the BH4 cycle (which breaks down catecholamines via MAO and COMT enzymes) operates in tandem with the methylation cycle. Many patients have polymorphisms affecting methylation and benefit from folate / B12 supplements which speed up this cycle and related cycles. The methylation cycle also operates in tandem with the trans-sulfuration cycle whereby homocysteine is converted into gluathione – our major detox / clean up substance. The reduced glutathione then requires NADP (from NAD) to recycle it into its active form where it can clean up free radicals produced by energy production, etc. If the reduced form of glutathione builds up (not enough NAD / FAD to recycle it), my understanding is that it is excreted in urine and glutathione levels fall in the cells, thereby further increasing vulnerability to oxidative stress. A potential source of a vicious cycle and a good argument for pacing.
Estrogen is also a COMT inhibitor and obviously this illness proportionately affects more women than men.
However, all of this said, I once read a pubmed paper that said 24 hour urine concentrations of vanillymandelic acid (the breakdown product of NE) in ME/CFS patients was the same as controls, suggesting NE levels aren’t higher in patients. Unfortunately after searching I can’t find the paper again though. Also, one would imagine that high NE would result in high blood pressure but most patients have low blood volume / blood pressure.
Maybe in some patients, the beta adrenergic receptors are subject to auto-immune damage or become resistant to NE, and as a result NE levels increase to compensate (thereby blocking acetylcholine production and inhibiting the parasympathetic nervous system) but renin production triggered by the beta adrenergic receptors remains low so the overall effect is low blood volume despite the high NE?
Strange illness.
Hi debsw,
Thx for your positive and insightful comments. As massive efforts of Issie and me proceeded these writings it feels good to know they were not in vein.
I’ll split my comments for ease of reading.
High sensitive people likely have faster firing neurons making more connections with each sensory input to other parts of the brain. Both should result in more extracellular glutamate and increased energy consumption.
Extracellular glutamate is excitotoxic under hypoxia when it is produced in too high amounts. Increased energy consumption of the brain easily leads to more ROS formation.
People with easy strong dopamine and nor-adrenaline spikes IMO also have a faster reacting brain with higher chances of being highly sensitive. With it, they may find it more difficult to remain “thought and feeling free” having a brain that is always “turned on”.
When overloaded, this increased accuracy of perception IMO easily degrades to uncoordinated massive brain firing leaving one confused.
I must add however that I feel that a “correct sized” increase in dopamine seems to get my brain remarkably focused and able to weed these continuous present stray thoughts out.
We are looking into methylation and BH4 too. That however is one of these topics that I have to reread again and again in order to slowly gain understanding.
Having a basic understanding of what each part does I can do. Understanding the complex interaction between several of these parts affecting each other however is very challenging to me.
I can get small improvements by doing basic changes concerning methylation. With Issie who has plenty of genetic problems, those backfire very easily likely due to the complex interaction of defects in these pathways. That learned us we are not ready to out what we try and do in this regard, as it could create plenty of havoc among patients trying to repeat 1 on 1 what we do.
I too thought for a long time we lacked the energy to recycle glutathione. It however gets a bit more complex.
I believe that during crashes we are hit badly by local hypoxia. That in turn increases glycolysis a lot. Glycolysis produces plenty of NADH.
NADH can create multiple ATP molecules *IF* the Krebbs cycle has sufficient access to oxygen. As during hypoxia NADH production is increased *a lot* and oxygen availability drops *a lot*, the mitochondria risk to be overwhelmed by NADH.
An abundance of high energy molecules like NADH may sound like ME heaven, but those molecules are unstable when too many of them pile up. When they pile up too much, they “discharge” their energy at high rate by creating plenty of ROS. That is *not* a good thing at all!
There are enzymes that can convert NADH to NADPH and it seems to happen at high speed during anaerobic metabolism. That would be a great source of NADPH for glutathione recycling.
However, it is the combined amount of NADH and NADPH that creates plenty of toxic ROS when it rises too high.
Glycolysis generates plenty of NADH during the combo exertion and local hypoxia. The conversion of pyruvate to lactate can reduce the amounts of excess NADH but will cause a strong surge in lactic acid. This acidity rise must be kept in check and this limits how fast the body can build down NADH. So the body seems to turn to very quick conversion in NADPH.
However, converting a big pile of NADH to a big pile of NADPH doesn’t solve anything. The pile could be decreased a bit by recycling glutathione but this doesn’t seem to be sufficient.
Rather, research points to the body to “switch on” a pathway to quickly reduce NADPH quite a bit *below* rest levels: fat synthesis from pyruvate, glutamate and NADPH. That reduces both excess pyruvate, extracellular glutamate and NADH/NADPH toxicity in the brain.
In the liver the same happens with excess pyruvate and NADH/NADPH. Frequent partial hypoxia hence can result in weight gain and non-alcoholic fatty liver disease. Acetaldehyde and high fructose consumption add to both.
Research studies show that this fatty acid synthesis during hypoxia protects the brain against hypoxic damage. It lowers NADPH so much despite a high production of it (from conversion of excess NADH to NADPH) that glutathione recycling speed drops.
Note that this fast fatty acid building is a safety mechanism, not something to block. If anything, the cause of this combo hypoxia and overexertion needs to be reduced.
L-cysteine (and several molecules made of cysteine), one of the building blocks of glutathione, reacts fairly well with acetaldehyde. This can both remove acetaldehyde and reduce cysteine availability for glutathione. I do however not estimate this a main driver for our reduced glutathione levels.
Note that using L-cysteine or NAC can be tricky for patients with certain mutations so don’t rush in with it yet.
This paper titled “Biochemical characterization of the catecholaldehyde reactivity of L-carnosine and its therapeutic potential in human myocardium” however points to a more likely problem:
“glutathione (GSH) showed essentially no reactivity towards DOPAL. In contrast, GSH readily reacted with the lipid peroxidation product 4-hydroxy-2-nonenal (4HNE)”
4HNE is another very nasty product that is created when massive oxidative stress attacks cell walls, just like acetaldehyde is created that way. I did not mention it before as it is nasty but offers no obvious route to the all important vicious circle needed for explaining why we *remain* ill.
The important thing here is: when massive ROS produces high amounts of acetaldehyde inside the cells, high amounts of nasty 4HNE will be produced too. That reacts with glutathione.
An interesting summary is found in a paper titled “Glutathione Dependent Metabolism and Detoxification of 4-hydroxy-2-nonenal”
“The magnitude of the sensitization to 4HNE toxicity caused by GSH depletion was similar to the magnitude of the reduction in the ability of cells to metabolize 4HNE. These results support the hypothesis that GSH and GST provide a biologically significant pathway for protection against aldehydic by-products of lipid peroxidation.”
As 4HNE can create *permanent* damage this product needs to detoxified well. Papers indicate glutathione can do this fast. There are many papers on the topic but I failed to find a single one that discussed recycling the glutathione formed by this specific reaction.
That indicates that this reaction may destroy the used glutathione in the process and reduce glutathione to a use-once-molecule versus a use–and-recycle-a-thousand-times molecule for scavenging ROS.
=> So I estimate the need to reduce combined NADH/NADPH during hypoxia to be the likely cause for increased oxidized glutathione versus reduced glutathione.
=> I estimate high lipid peroxidation producing 4HNE (and acetaldehyde) to be the cause of low overall glutathione levels.
MAO and COMT enzymes are both needed to break down dopamine and nor-adrenaline. Their *ratio* however seems to be very important.
MAO is the first step to convert dopamine to very toxic DOPAL. DOPAL is said to be !1000! times more toxic then dopamine according to a research paper. So it piling up is a big problem.
Now how high it piles up is dependent on the ratio of reaction speeds of each step.
Consider for example a border control with 3 check points (and plenty of place in between for people to accumulate).
Checkpoint A: state your nationality and walk through.
Checkpoint B: fill out a detailed 10 page questionnaire and discuss all of it with a customs officer.
Checkpoint C: open up your suit case and have every single item checked
It’s easy to see if you have one “line” where those checkpoint are organized in order A -> B -> C that the masses will not accumulate in front of (first) checkpoint A but will accumulate in front of checkpoints B and C.
If you have a second line organized in order B -> C -> A then people will accumulate in front of (first) checkpoint B and C but not in front of checkpoint A.
If you have both custom lines, more people will tend to chose the first line as that seems to go quicker. So which step happens first and how fast that happens will determine in a large part how much “traffic” each path sees and where the people accumulate.
When translating that to dopamine degradation:
Having fast MAO versus slow COMT will send more dopamine for degradation to the COMT (DOPAL) pathway *and* a strong accumulation will happen at the “DOPAL checkpoint”.
If you have slower MAO versus fast COMT, more dopamine will take the more benign 3-methoxytyramine pathway and less DOPAL will be generated and it will be faster detoxed decreasing DOPAL build up a lot.
NOTE: don’t self experiment with MAO, DAO and COMT related drugs! Things can get very tricky very fast!
From the paper titled “Comparison of Monoamine Oxidase Inhibitors inDecreasing Production of the Autotoxic Dopamine Metabolite3,4-Dihydroxyphenylacetaldehyde in PC12 Cells”
“Combined MAO-A/B inhibitiondecreases DOPAL production in rat pheochromocytoma PC12cells”
But as said: DON’T play by yourself with MAO inhibitors! MAO does more vital things then just this!
“24 hour urine concentrations of vanillymandelic acid (the breakdown product of NE) in ME/CFS patients was the same as controls, suggesting NE levels aren’t higher in patients.”
=> I suspect several factors:
* It may swing more from low to very high, creating more havoc.
* Both dopamine and NE are very reactive chemicals. Some of their metabolites are even more reactive. Many lipid peroxidation products like acetaldehyde, 4HNE, methylglyoxal are also very reactive. Some of those react with the dopamine / NE related chemicals creating different waste products then HVA that likely were not measured in that paper.
* Dopamine, NE and many of their metabolites also act as a sort of anti-oxidant. With ME patients likely having plenty of ROS during dopamine / NE surges much of it might react away in this way again ending up in different final degradation products then HVA.
I just had a hunch and looked it up: HVA is an anti-oxidant by itself.
=> Under the conditions of high oxidative stress measuring 24h urine HVA alone would be a poor measure of determining NE levels in ME patients!
See a paper titled “A Simple Assay for the Measurement of Plasma Antioxidant Status Using Spontaneous Autoxidation of Homovanillic Acid”
Saying “In addition to this scavenging effect, the kinetics of HVA autoxidation, restarting after the delay, reflects the ability of the plasma antioxidants to inhibit the ROS-triggered autoxidation.”
That describes that the auto-oxidation of HVA stopped in their experiment due to reduction in ROS if they added other anti-oxidants to the sample. So when other anti-oxidants are low, HVA acts as an anti-oxidant. It’s fast enough an anti-oxidant for its content to reduce fast enough under the condition of high oxidative stress to be usable as a marker of oxidative stress itself.
I don’t know what your paper did asses, but details seem to matter!
Oops, HVA is a breakdown product of dopamine. You referred to vanillymandelic acid (the breakdown product of NE).
But that also is an anti-oxidant in some form.
See the paper titled “Antibody-Catalyzed Decarboxylative Oxidation of Vanillylmandelic Acid”
They use some chemical too as their purpose is industrial, but notice the use of anti-bodies to speed up the reaction. In auto-immune diseases anti-bodies may be high.
An old paper titled “THE ENZYMATIC OXIDATIONOF p-HYDROXYMANDELIC ACID TO p-HYDROXYBENZOIC ACID” says:
“On the basis of structural similarity it seemed likely that the oxidation of p-hydroxymandelate might proceed in a manner analogous to that of mandelate.The degradation of the latter compound to benzoate with the formation of benzoylformate and benzaldehyde as intermediates”
That is done by some sort of bug. Both mandelate and hydroxymandelate are common waste products of NE.
=> So these waste products can be oxidized in biological only reactions. That would reduce their markers under the condition of heavy oxidative stress.
In my search I also did find a paper with title “Oxidative and Nitrative Protein Modifications in Parkinson’s Disease” with a table linking increased lipid peroxidation to decreased GSH (glutathione) (layout of the table screwed up with copy paste)
“Increased lipid peroxidation, protein carbonyls and 3NTs in total protein Decreased GSH, increased oxidative stress”
That further supports the idea of excessive oxidative stress and lipid peroxidation having a role in decreased glutathione as expected in a comment above this one.
The paper also contains *FOUR* different pathways (likely related to what happens in ME) for complex I inhibition in the table, with it inhibiting mitochondria.
In the text itself there is a fifth “We previously demonstrated in vitro that decreased glutathione levels resulted in selective inhibition of complex I activity.”
@debsw, thank you for your comments. I’m sure Dejurgen will get back to you tomorrow.
We are looking into the issues with methylation and do feel there is a connection. We are trying to not put everything out, all at once. As you said it is complex and deep and we are trying to not overwhelm or go to technical. (Though Dejurgen sure can go very deep into science and then it may not be understood. I love the science, but it and he can go zoom, zoom. Right over ones head.)
I personally do have issues genetically with BH4, but not COMT or MAO. And I do have very high NE and low blood volume. And nearly non-existent renin and aldosterone. I have higher blood pressure (actually variable blood pressure). But I also have Hyperadrendic POTS, ME and a whole list of other DXs.
We know everyone is unique and what may be true for one subset, may be different for another. And what will work for me with my wonky genes may not work for someone else. Dejurgen and I are addressing similar things but we are having to do it through different pathways to get to a common goal. What he can use, I can not. But we are finding ways to work around the differences.
What you speak of has merit and we are finding connections in those areas. Thank you so much for your comments and observations. We all are searching for our “purple bandaids”! Let us find them!
Issie
I thought the Norwegians Drs Mella Add Fluge had Tried to do an entranercet drug trial but abandoned it as it was having too many adverse effects. Or was that study purely of the severely affected where sensitivity to medication might be stronger? I remember being disappointed as I’d been trying to get hold of the drug to try for years.
I wonder why some people get terrible pain and Some none?
I’m bedridden and apparently the typical presentation is constant full body pain whereas I get none although I do get strong constant discomfort from untreatable spasm and have very distressing cognitive issues akin to head injury type dysfunction and confusion. Pain can be a Bodily protective mechanism often and I wonder if those of us fortunate enough not to have it are also without the shielding, protective mechanism So it’s actually just a fault In a few individuals ….
Too high DOPAL creates massive ROS inside the cells and more so inside the mitochondria of these cells. As mentioned above that happens most in the “dopaminergic neurons” including the “primitive brain” parts like the hypothalamus and PVN part and in the motor neurons.
The PVN part, as Angus Mackay explained, filters senses and regulates the HPA axis. That and other parts filter pain inputs and directly modulate pain sensation. That and other parts filter sensory inputs like sound, vision,… regulating or failing to block the common sensory overload.
Limb and muscle fatigue and weakness is directly related to the affected very dopaminergic motor neurons.
Now both too high DOPAL and too high ROS have a strong inhibiting effect on mitochondria.
That will force these local brain regions to turn to anaerobic metabolism a lot more. That will turn plenty of NAD+ to NADH, leaving very low amounts of NAD+. That NAD+ however is required for the local ALDH enzymes in that part of the brain to do their work. Those local enzymes are needed as a first line of defense in order to locally decrease excessive DOPAL levels. So once too much DOPAL starts too inhibit the dopaminergic neurons, DOPAL will slow its own detoxification by depleting NAD+ needed by the ALDH enzymes that are key to detox DOPAL.
That is a first part of delayed building down of the nasty and toxic local situation. But it gets worse, a lot worse.
Part of the likely very high amounts of ROS created by the high amounts of DOPAL building up inside the mitochondria and cells will attack the cell walls and create massive amounts of acetaldehyde inside the mitochondria and cells. IIRC one reactive oxygen species attacking the cell walls can create a chain reaction damaging hundreds of cell wall molecules and creating hundreds of molecules of acetaldehyde and some other equally nefarious chemicals. I have to check that info however but ROS attacking cell walls for sure will create massive acetaldehyde.
Acetaldehyde in itself is a very toxic substance in too high concentrations and it is believed to be a main part in alcohol toxicity during alcohol abuse. Note that drinking some modest amount of alcohol for ME patients who already produce too much acetaldehyde this way may make symptoms worse as many of us report.
Both ROS and acetaldehyde, once “escaping” to the main blood flow, are known to stiffen RBC making them more inflexible. This will, over time, further degrade oxygenation and increase chances of getting in a deep hypoxic condition near the smallest capillaries helping to entrench this situation.
Getting back to the topic at hand, acetaldehyde messes with plenty of chemical processes and the mitochondria too. But there are other aspects of acetaldehyde:
Acetaldehyde in too high concentrations triggers… dopamine release. So if DOPAL, a waste product of dopamine is getting too high, there is plenty of chance for excessive ROS to be generated in dopaminergic neurons. This risks to both damage cell walls and create plenty of acetaldehyde. That in turn risks to create a spike in dopamine, creating even more “raw materials” to build this VERY toxic DOPAL from. If so, we are in for one BAD storm.
Oh dear I am not looking forward to this but I will give you my own experiences with endogenous acetaldehyde as a cause for some of it :
-The acetaldehyde literally creates pain in the body and that local pain is as real as can be. It is “there”. When I was very ill I had severe pain literally everywhere and I my brain was very “vigilant” and “aware” of it. Makes sense does it not that your brain would do that right ?
– The acetaldehyde makes you very touch sensitive it seems to “excite” all the nerves. I could also not stand certain things touching me or handle uncomfortable clothes. After coming home from something the first thing I would do was change clothes I could bear only underpants, a loose fitting T-shirt and jogging pants, nothing more. And NO SOCKS. And only slippers. And constantly re-adjusting blankets for sleeping and such because it bothered me and did not feel right.
– The acetaldehyde makes the brain completely “hyper” to literally everything. It works as an amplifier everything that normally would not be noticed is now being noticed as a problem so you become extremely aware of it. It can make you hyper-aware and highly anxious even if you yourself don’t see any psychological reason for it and don’t understand why it is happening. It can even create real fear for something, I was literally “fearful” for things that never bothered me before. This has got to do with that flight or fight response, it is how the brain works under the influence of the acetaldehyde it acts as a real “drug” on it.
– There is absolutely an over-emotionality aspect to ME / CFS, that is how some people come across / appear to the outsider or shortsighted doctor / researcher. And people even notice it themselves. But it isn’t psychological, it just LOOKS that way. The acetaldehyde does that to you and you can’t help it. You can only compensate so much for it with willpower but in the end the drug-effect is still there and it still influences you in your actions and thinking.
– The acetaldehyde is a strong irritant to all living tissues and it effects how they function. All organs in the body don’t like it very much. Maybe this can create mini-inflammations or something like it.
Cort is always doing such fantastic work describing what is happening with ME / CFS research and I find it always interesting to read and please don’t see it as non-constructive criticism or negativity what I am about to say ( it is all understandable) but :
The thinking behind some of these researchers and what they are researching and what they think might be a cause, or the ideas that people with ME / CFS come up with themselves sometimes, just don’t seem to ever explain all the wide-ranging whole-body symptoms people with ME / CFS have.
Ah dejurgen just posted something about it mentioning acetaldehyde right before me that is funny ! What I would like to add is that how exactly all the biological processes happen in the body under the influence of acetaldehyde is still quite a mystery to me too…. I only know that the acetaldehyde seems to be the main cause of all my ME / CFS problems. It just “does that”….
Thanks buddy, you sure as hell influenced my ideas!
You were first with the idea of acetaldehyde being potentially really important. Thanks for your lonely fight to promote your ideas! I could not link it to other ideas I had at the time though.
Later on I came through a different route to the same conclusion: acetaldehyde may be one of those chemicals playing more then a big role into many cases of ME/CFS and sibling diseases.
I do mention “through a different route” not to compete with AcH influenced but to stress that several observations seem to point to the same chemical. I still can’t say that it is vital in a majority of cases but it has the real chance to play a nefarious role in a large subset of patients.
I hope our combined effort in this, AcH influenced, Issie and I not only helped pointing to acetaldehyde but also to point to potential biochemical pathways (on top of pathogen generated acetaldehyde).
Acetaldehyde spikes creating more DOPAL has some insidious effects.
Extra dopamine gives the feeling of both energy and strength in the muscle and brain. It is a chemical of well being and addiction.
So at the onset of this “toxic storm”, we get hypothesized partial and local (near the finest capillaries) hypoxia. More DOPAL production and slower DOPAL detoxification. This in turn creates more ROS and acetaldehyde.
This *should* inhibit the mitochondria straight away and cause a strong sense of fatigue. But, this strong increase in acetaldehyde creates a spike in dopamine. This in turn increases the “raw materials” available to create (nor-)adrenaline a lot. So we potentially not only have a strong increase in dopamine but also a substantial increase in nor-adrenaline and adrenaline.
Dopamine creates energy in brain and limbs and a feeling of well being. It is also the hormone of addiction that lowers our estimation of risks and their bad affects on our lives.
(Nor-)adrenaline is the hormone that can allow frail old people to believe they can lift a car (and sometimes even actually succeed at it) when they believe they can save their grand child blocked under it. It gives a massive boost in strength and (depending on individual characteristics: fight or flight?) potentially a massive boost in mental focus.
Now imagine overdoing it creating a local anaerobe functioning in the brain. That is in fact a trigger point to slow down and pace urgently. In healthy people it does likely create a surge in DOPAL and ROS big enough to allow them to feel fatigued and stop and rest. But with us (due to certain pre-conditions different compared to healthy controls) that creates this surge in toxic DOPAL, ROS and acetaldehyde big enough to trigger a big “second wave” of (feeling of) energy due to a strong surge in dopamine and (nor)-adrenaline.
This “second wave” cancels and or completely overrides the feeling of fatigue risking to give us the feeling to be at our best and have an upswing in energy. Combine that with the hypothesized surge in dopamine making us blind for risk and the surge in (nor-adrenaline) making us blind to the fact that we are overexerting a lot and you have a near perfect recipe for overdoing it time and again even if we are smart enough to know it happens and takes us by surprise time and again.
We may feel at our best thanks to overexerting for a while thanks to this mechanism and be unaware we are close to being at our worst! That sure could trigger us to delve each time way to deep into our reserves and prepare us for a nasty crash! Actually, one of the things I believe that “prepared” me for this (in my case gradual onset) disease is the fact that I learned about this “second wind” when I “had to” study twice as hard in order to not lose my year after being hit by mono/EBV during my university studies.
I learned when I was really fatigued and barely could go on studying, if I just pushed even a bit deeper I got this second wind giving me *more* energy then when being well rested and it even felt like a slight touch of deep mental focus and insights. I sadly learned to use and abuse this mechanism on a very regular basis the following years as it offered me an unknown feeling of health and mental and physical energy during several years. But the cost was very very high I fear.
Overriding this early inhibition and safety mechanism (strong sense of fatigue and need to rest) with this surge in dopamine and (nor-)adrenaline time and again IMO allows the toxic stuff to build up far beyond what is safe and sound each time, creating plenty and hard to detox waste weakening our bodies with each following hit.
Where this “second wind” gives us a strong sense of more well being and energy at a time we should rest, it also potentially creates a first phase in PEM: overdoing it but not feeling it yet due to the flood of hormones masking this feeling.
So when do we start to feel the crash? There are likely several parts in this.
One part seems to be the amounts of toxic DOPAL (and a few similar VERY toxic chemicals in its wake) pilling up. When we keep overdoing it and stressing our mitochondria far beyond what is safe (by having among others parts of our brain going too long in deep hypoxia and anaerobe functioning), DOPAL should build up.
That in turn will increase toxic acetaldehyde levels and for some time even further increase dopamine production. However, at some point the inhibiting effects of toxic amounts of DOPAL, ROS and acetaldehyde should outweigh the activating and well being effects of increasing levels of dopamine as the toxic chemicals likely will increase faster then the surge in dopamine and (nor-)adrenaline can follow (and that *VERY* likely is a vital protection mechanism).
At that point, mitochondrial inhibition will start too outweigh mitochondrial activation by the combo surge in dopamine and (nor-)adrenaline.
At the same time, anaerobic waste products will have kept pilling up likely resulting in a large spike of waste products like too high lactate and pyruvate (and alanine) levels in the brain that needs to be detoxified by the liver. But for each ATP gained by the hypoxic brain regions by anaerobe metabolism, the liver needs three ore more ATP depending on the exact detoxification pathway. That increases oxygen and blood flow demand by the liver to very high levels.
That not only increases demand for breathing a lot, but also diverts blood flow from all parts of the body to the liver as energy needs likely are massive over there. That effect of diverting is felt even stronger in us as our blood volumes and flow already is reduced, so each amount of redirecting blood flow towards the liver has a stronger reduction of blood flow to other organs like the digestive tract and brain compared to what happens in healthy individuals… …increasing hypoxic and anaerobic functioning (and inhibition to save oxygen) further strengthening the vicious circle and prolonging normalization of oxygenation a lot compared to healthy controls.
Once again, it gets worse as both ROS and acetaldehyde can not only damage RBC but also bind to hemoglobin (and only release slowly from them), decreasing the RBC capacity to carry oxygen… …further decreasing oxygenation to the entire body for a prolonged time after this crash.
There is another element in this. ALDH enzymes need NAD+ and with it indirectly oxygen. In the first phase of the crash it was hypothesized that the local activity of ALDH in the dopaminergic neurons was reduced by lack of NAD+. That offloads a bigger part of the detoxification of DOPAL to the liver. That very likely goes slower as the DOPAL needs to travel from the inside of the mitochondria towards the liver. But when the entire body gets hit and plenty of other processes requiring urgent detoxification by the liver using plenty of oxygen and NAD+… even detoxification of DOPAL (and some other very toxic chemicals) in the liver by ALDH enzymes risks to slow down… …a deep crash in the making?
As often, it even gets worse. When there is enough DOPAL to react at fairly high speed with dopamine itself, its creates a chemical known as tetrahydropapaveroline. That chemical inhibits the uptake of dopamine by the brain.
So when we are “far enough” in the crash for plenty of DOPAL to be present and react with increased (due to this “second wind” caused by high acetaldehyde levels) levels of dopamine, plenty of toxic tetrahydropapaveroline is produced. That inhibits the uptake of dopamine by the brain (and dopaminergic neurons are located in the brain too).
That likely is an essential safety mechanism but one with a nasty taste: suddenly dopamine levels in the dopaminergic neurons start to drop, potentially and likely deep below the levels at rest.
If so, we now have the very strong mitochondria inhibiting effects of DOPAL and ROS, the interfering chemicals like acetaldehyde AND reduced levels of dopamine (in the dopaminergic neurons) (and potentially later in its wake reduced levels of (nor-)adrenaline) all combining to inhibit dopaminergic neurons in a “total” fashion: PEM potentially hits at its hardest here.
That plus the derailed blood flow, damaged RBC with bound ROS and acetaldehyde, plenty of toxic waste to be cleaned up and later a mechanism similar to reperfusion injury creating massive inflammation “should do”.
Note that during “real” ischemia the reperfusion injury is often said to be more damaging then the ischemia itself. Reperfusion injury is the injury caused by massive inflammation after deep hypoxia when oxygenation and blood flow restores.
Now where does COMT comes into play? We already mentioned its role in dopamine and (nor-)adrenaline metabolism. But the COMT genes and enzymes are strongly related to methylation.
And methylation of DNA is strongly related to not only addiction, but also to high levels of acetaldehyde. Actually, it is believed that changes in methylation due to high acetaldehyde levels play an important role in addiction.
Let me stress we are NOT addicts, but a very similar biochemical mechanism seems to hit us.
It is not a straightforward “all DNA is more or less methylated” thing as that depends on specific tissue. Let us suffice to say here that over time plenty of DNA is “methylated wrong” or better said “different” compared to healthy controls.
Restoring that balance requires COMT to be active as this “wrong methylation” has build up over vast amounts of DNA. This leaves the option of COMT enzymes to be “pre-occupied” by trying to restore balance of DNA methylation. If so, that would leave less COMT enzymes available for doing the “usual things” like playing a role in dopamine and (nor-)adrenaline metabolism.
In plain speak: it likely would appear as lower COMT activity when it comes to the speed of COMT in dopamine and (nor-)adrenaline metabolism.
If so, this in fact may negate the “requirement” for COMT gene polymorphisms for COMT enzymes and speed to be impacted. Having methylation problems (for example due to CBS polymorphisms) or acetaldehyde induced “wrong” DNA methylation might act very similar to COMT polymorphisms.
Having several of these together might increase the risk to get into ME and make getting out of it even more difficult in this hypothesis.
Writing this I also just found “Acetaldehyde Accelerates HCV-induced Impairment of Innate Immunity by Suppressing Methylation Reactions in Liver Cells”
@Veronique:
IIRC research points to DNA methylation playing an important role in things like brain development under alcohol consumption of the mother during pregnancy and into how prolonged periods of stress impact the developing brain.
That would open up a potential route to the impact of mothers with high levels of stress and or ME onto the developing brain of the unborn child, and the impact of early stressful events during childhood and would also have a link to autism. I’ll leave that part to your expertise ;-).
Thanks for all of you who managed to read this far (don’t feel bad if this is way too much of a task),
Issie and dejurgen
I do leave one very new and “potentially interesting” new find here. But I warn that there are still some issues with it:
Half of the estrogens (estrogen is a collection of chemicals rather then a single chemical) are Wikipedia(Catechol_estrogen).
From the Wikipedia site I quote “Under poor conditions of inactivation by phase II enzymes, catechol estrogens can undergo oxidation to reactive quinones and semiquinones”
Many reactive quinones and semiquinones are highly inflammatory.
In plain speak: they depend on the COMT enzymes for degradation. That makes that (adult) females, with generally far higher levels of estrogen then men (and young girls), “need” to make more use of their COMT enzymes.
That would potentially open up a partial explanation why adult females are extra prone to this type of diseases. The reasoning here is that if their COMT enzymes are already faltering, they need to use it for one big extra task (estrogen degradation) that men and young girls have to do a lot less.
Build up of some of these estrogens and estrogen degradation products also interferes with dopamine, nor-adrenaline and adrenaline metabolism.
Note however: this idea at first sight seems to conflict with the observation that pregnant women often get a strong improvement while having more estrogen.
It also conflicts with many menopause women getting worse ME symptoms and experiencing benefits on HRT.
Issie and I are working on analyzing the situation. It appears estrogen has several protective effects on processes that might influence ME vulnerability. Also, estrogen itself affects COMT genes and enzyme expression differently in different parts of the body including in the brain.
On top of that, exact estrogen chemicals compostition and changes in it may effect how it influences ME. It seems for example (preliminary finding) that mainly estriol increases during pregnancy and that seems (we need to look deeper into it) to not be a COMT dependent estrogen chemical.
It thus may be that both composition and amount best are optimal rather then low or high.
As said, interesting for sure but preliminary!
couple of things : the mentioning in the blog post “pain is being brought to the body by the brain” I can confirm as maybe a part of the problem. I wrote about that on my website that this is also happening and that there is a back-feed loop that amplifies things so that might be interesting to read about for people. When I was really ill I could generate a thought about falling and I would have a whole body fall-reflex shudder as a result. It was actually funny to do that ! When I had a thought about something that could generate pain or also when watching a youtube clip where someone fell / smacked on the ground it gave me a jolt of pain myself. I became a bit of an “empath” hmmpff… my whole brain acted hyper / over-emotional to everything you get crazy effects. But I would still strongly emphasize that firstly the physical pain is absolutely real, these days being less ill those weird brain reacting effects are a lot less but I still have physical pain it is just less.
I believe that many a person with strange complaints and overlapping symptoms has this acetaldehyde problem instead and it is involved in it as a comorbidity or is even the actual cause of it. So they think “I have Lyme disease” or “I have an auto-immune disease” but instead they have this problem. It brings out all kinds of weird symptoms just like it. Everyone is a bit different and this general problem as the first cause brings out your own personal health vulnerabilities.
The endogenous acetaldehyde can only be produced by a biological organism, a biological mechanism or a combination of both. We are stuck with that. But given how my illness developed with the gut issues completely linear with the acetaldehyde production and given that I managed to make it all less with antimicrobial stuff and low-carb / keto / carnivore diet I think we are having a ” bug” problem….
>dejurgen you mentioning the RBC and hemoglobin : yes I believe that is definately happening and very important. It is involved in the oxygen exchange / shortness of breath problems. It might also be very important for measuring acetaldehyde because the acetaldehyde is for a large part bound to the red blood cells according to alcohol research. I will gather some interesting links that might interest you and post them in my acetaldehyde topic this weekend.
@AcH influenced, I can sooooo relate to your feelings and am myself super sensitive. Dejurgen and I are both making big discovery in this field. And us both addressing all that he is writing about, is giving us both considerable improvements. We are both feeling the connections we are making are HUGE!
Slowly, he is releasing what we are connecting. We do hope to get it all in one place. But we don’t want to not “TELL” should it help others. It can go very deep and be complex. But dejurgen is doing a wonderful job of explaining it simple. You should see how deep he can get with it and that would definitely go zoom, zoom! We have both done so much reading, talking, connecting in the last year. And are ready to start sharing. Hope it makes a difference!
Thanks for putting your experience out there. It gave us something more to reaffirm what we were finding. Looking forward to what you post this weekend. Just don’t crash yourself.
I will post the links about the nature of acetaldehyde and that they still, to this day, still have not figured it out what effects the acetaldehyde exactly has on the brain. It is largely unknown ! It is weird, weird, weird stuff… It does strange things to the brain. It took me 3 years to find out that I was “half-drunk” and I was an extreme case so it is no surprise that if someone else would happen to have this at a lower level they would never make the connection that it could have anything to do with their symptoms and just live with it not knowing. And why would they ? Who on earth knows what acetaldehyde on its own feels like ? There are no examples (except for alcoholics on the Antabuse who take small sips of their alcohol but that is a different category).
We will get there in the end I think. I can fully imagine people just don’t know what to think of it.
AcH influenced, dejurgen and issie,
I spent ages trying to figure out how/why I could feel drunk, when I wasn’t. I even bought a travel breathalyser kit to test myself – negative result.
I linked it to eating too much fructose. Robert H Lustig, who has a palpable loathing of all things sugar, and especially fructose related, wrote an article Fructose – It’s “Alcohol without the Buzz”. He mentions acetaldehyde.
Fructose, especially from corn (like modified maize starch) – is my arch nemesis – my kryptonite. At higher levels it seems to sedate me and I could lie completely motionless, though ‘awake’ for hours. The most dangerous aspect would be it’s ability to affect my breathing – which would become very, very slow. I would also have to consciously breath in and out, in and out. If I stopped doing this, I stopped breathing.
Obviously that sounds completely ridiculous to anyone else…
Other symptoms I would experience with fructose would be – nausea, flattened mood, memory issues, inability to do maths and a need to pee a lot.
Anyway, the fructose issue was the first I encountered in 2007 and it persists to this day. However I generally do not eat it. No fruit, no higher fructose vegetables, no corn products etc
And dejurgen/issue I’m not ignoring your detailed comments – I honestly just can’t keep up! I read the Tate and Mackay blog by Cort, made my comment and then saw your comments dejurgen and thought Hmm, I’ll come back to that when I feel a bit more fortified. However the fortification never arrived and then we moved on… Ah!
I don’t know if it’s possible for one of you to do a really simple summary for people like me? Tracey ?
Morning Tracey Anne,
Thanks for replying! It’s heart warming that this really big effort doesn’t go lost. I do know it’s plenty of dense information, a wall of it as to speak. Don’t worry, when I was at worst after reading 10 lines of text I got confused and letters started swirling over the page…
When drinking non-alcoholic beer I learned that I had half of the alcohol effect without the buzz. Wheat is bad for me, liquid wheat like alcohol free beer is worse and fructose rich food is plain disaster. Issie has it similar ways.
Almost being unable to breath for an entire night? Been there done that! That was when I probably had near no stress hormones anymore. I couldn’t get angry, couldn’t worry. I just could focus on trying to breath and make it through the night. Even then my chest barely moved. I wasn’t panicking in any way but with such low breathing survival wasn’t obvious. The next time it happened I felt confident I’d make it.
A summary? Normally I’d say it is already summarized a lot. The topic is big! But I’ll do an effort for you that will fit into a little idea I was thinking to post already.
When exerting and already digging deep in our reserves, it seems we are producing several toxins in high amount. One of them is this acetaldehyde chemical. That can oddly enough create a boost in dopamine. There is research about that.
If the boost in dopamine is big enough, we may feel a surge in mental and physical energy and well being, making us feel better then at rest. You can imagine that is very tricky as it will compel us to do more now we for once feel actually quite good.
Unfortunately that boost doesn’t protect us against further build up of dangerous toxins when we further exert ourselves. Over time, the nefarious effects of even higher amounts of toxins will overwhelm this boost in energizing hormones. Our energy levels will start to drop to normal levels.
By then we are already very deep into the danger zone. From then on, we’re in for a deep crash leaving us with a massive pile of toxins and few energizing hormones. Well pay for it for days to come: PEM!
The very simple yet difficult to follow idea / advice?
We need to pace more yet fail to pace in time over and over. We don’t feel the oncoming crash soon enough. But if this theory is right, we are already exceeding our limits when feeling at its best!
And feeling at our best, that we can estimate perfectly well! We crave for these moments. We recognize them in an instant. And we decide to take advantage of them.
So, we recognize them very well. Yet, we already are overdoing it? Then it’s time to stop and rest! Yes: stop and rest just seconds after we feel we past the peak!
That brings me to the very simple yet so hard to follow advice: make “leave the party at its best” *bit by bit* your life motto.
*bit by bit* = don’t go in head over heels changing your lifestyle at once.
It may make no sense at all when first hearing it, wasting all that opportunity to do more when feeling at your best. But you aren’t! You are showered with hormones pumping you up and trying to save you.
Actually, they’re perfectly usable for better recovery if you just stop and rest. No more crash overdo crash overdo crash overdo continuous cycle, isn’t that worth more then just 5 or even 30 minutes more of joy? Slowly feeling stronger and recovering bit by bit over time, isn’t that better then feeling well for 5 to 30 minutes before crashing for days?
As a bonus, during the following days you’ll live on the memory of the joy you had at the top of the fun, “when leaving the part”. You’ll thrive by it and the memory will give you a slow and low and far safer steady stream of dopamine and oxytocin giving you that small extra boost.
It wont just heal you, but this simple motto may be “pacing 2.0”.
@Tracey Anne, thanks for your comments. I’m the same with fruit, grains and especially corn too. Those things had to just go away. Limiting sugars and going low glycemic even on vegetables. Carrots are to sweet for me. A few in a soup are fine or a few grated in a salad, but to eat a serving of them…..can’t do. One of my worst reactions.
You should see how technical and sciencey dejurgen can go……he has simplified it a lot. Even with my understanding of most of the medical and science of it…..I ask him to “dumb that down” for me. LOL. When I first got started down the path of medicine and science, I had to look up individual words……so I know the frustration of wanting to understand and not being able to. Keep reading, it will just click one day. But then when brain fog occurs……just give it a day or two and come back. When I’m like that……I can’t understand even the simple things. I’ll tell dejurgen “I’m brain dead today…..explain this to me”. And he will. Usually he will be up when I’m down and vica versa.
I’m glad you are getting benefit from what we try to do…..that is our goal…..to find “purple bandaids” and help others and therefore help ourselves too!
Issie
Thanks dejurgen and issie
That’s great – I can understand that… ?
@ Tracey Anne : Oh that is really really good that you posted your experiences so now there are already more people who can make themselves feel “drunk”-like. Haha I really like it that you tried testing yourself with the alcohol tester ! You actually tried finding out about a cause for you feeling drunk well that is really telling something is it not ?
I had my blood tested for both alcohol and acetaldehyde . My alcohol (ethanol) was undetectable so that was not the cause of the drunkenness but the acetaldehyde was sky-high. I can absolutely confirm that it is possible that you can get a lot of drunk-like effects on the acetaldehyde alone. It is really the cause of all these brain dysfunctioning things in a lot of people.
The drunk-like feelings and brain fog and such have actually already been mentioned and discussed by other people sometimes related with people trying out low-carb / keto no sugar and it often improves ( I can confirm that that is part of solving / improving the problem) . The whole thing is actually not rare at all I think but I believe that no one actually understands that these problems can be caused by the acetaldehyde. They just don’t know !
I have exactly the same problem with doing math, I have real problems even doing simple multiplications ! The strange thing is that rational thinking and such is still quite okay but executive functioning / tasks go down the drain…
I also had strange breathing effects like air- hunger and feeling very aware of my breathing / lung-feelings so I know exactly what someone means when they mention their breathing symptoms.
The interesting Robert Lustig paper https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3649103/ is also interesting for dejurgen I think it also mentions ROS and such
@AcH influenced:
That indeed is an interesting paper! It discusses potential pathways on how it works in detail. I like that a lot.
Let me emphasis a few points related to our dopamine / acetaldehyde comments:
* Fructose spikes increase dopamine in the brain. As a result, the amount of dopamine receptors in the brain declines after long term high fructose use. That creates a dependency by needing more dopamine for normal functioning.
=> As long term frequent acetaldehyde spikes create dopamine spikes in the brain too, in between these spikes our dopamine response is lower then that of healthy people.
=> If the previously discussed ideas on the combo acetaldehyde / dopamine / noradrenaline would be correct, then after a crash we have a triple hit in both well being and functioning of those parts of the brain depending on dopamine for good functioning:
* increased toxic waste like DOPAL, ROS and other waste inhibits the dopamine depending neurons / mitochondria
* decreased activation of dopamine receptors versus healthy controls due to a decrease in dopamine receptors
* decreased uptake of dopamine by the mitochondria as tetrahydropalmatine inhibits the dopamine transporters
=> As dopamine plays a vital role in both motor neurons and key “primitive” brain areas like the PVN and VTA our motor functions, sensor filtering and hormone and temperature regulation becomes a total mess after a crash.
* Another interesting part is that acetaldehyde increases conversion of carbohydrates to fat. This statement is backed by research papers. That helps explain why *part* of us are hit by rapid weight gain when getting ME.
* A last interesting point I wish to mention is that high fructose levels create 7 times as much AGE and ROS compared to glucose. Both those products are pro-inflammatory.
The acetaldehyde/dopamine connection is interesting.
I found that if I hadn’t eaten anything sugary for a while my levels of ? would go down.
That would lull me into the false idea that, in fact, eating chocolate/cake/biscuits/cookies was just fine because eating something sugary was often okay the first time.
However on day 2 or 3 of my excited and happy little venture back into the tantalising sugar laden world, the sparkle would vanish and a dull, grey world would replace it.
Now I would feel tired, vague, apathetic and depressed. I would have a leaden brain and a headache. I would feel nauseous and have a horrible taste in my mouth. I’d need to lie down, my breathing would slow and I’d need to noticeably pee more.
I’m taking all that information from a small piece of paper that I wrote the list of consequences that occurred, if I ate too much sugar/fructose.
I wrote the list because I would forget how bad I felt, when I’d cut the sugar out and returned to ‘normal’.
After a few weeks it would be a distant memory – so I’d try a little something, be okay and think I was now ‘cured’ – have some more the next day and then realise (too late) that – no, unfortunately I wasn’t!
That cycle happened so often, I thought I’d write down just how bad I felt, to remind myself that – No, I could not repeatedly eat sugar/fructose…
I felt like I couldn’t process the fructose – it seemed to turn into poison. I wonder if that was the acetaldehyde?
@dejurgen it is good that you mentioned the possible acetaldehyde weight gain connection because I have read that too but had completely forgotten about that ! I believe that is most certainly the case with me. I think I have even noticed that in the sense that when really ill ( the most acetaldehyde) for the same amount of food I gained more weight more quickly than these days now I am somewhat better. Yeah I really think so. The pounds seemed to stack up more quickly but I don’t believe it has anything to do with age.
I could have missed it ? but you never mentioned Salsolinol wich is often mentioned with the acetaldehyde research and is the condensation product of dopamine and acetaldehyde. It has got to do with the opioid receptors and why they think acetaldehyde might be responsable for addictive behaviour. ( yes it is and more, it “rules” you…) Oh you find a library full of information about it in alcohol related research !
That idea of using notes is a smart one, if you follow it.
Acetaldehyde could be related to many of the symptoms, but many could as well be related to big portions of fructose reaching the big bowel.
There fructose would feed the gut bacteria that typically live on “slower fuel” like fibers. That could easily disrupt gut microbiome balance and create both inflammation and brain symptoms.
Many micro organisms in the big bowel can also produce a certain amount of acetaldehyde when fed with fructose.
The fructose would also put a heavy load on the liver, slowing detox of other toxins including acetaldehyde. According to the paper a fructose spike in the blood also forces large amounts of pyruvate onto the mitochondria, increasing reactive oxygen production. That in turn can produce acetaldehyde.
That are but some of the better candidates to explain what could have happened. In order to know more, one often has to observe the individual process many times in detail.
Thanks AcH Influenced. I never heard of salsolinol before. It seems to be another nasty product that has high toxicity, interferes with dopamine metabolism but it also interferes / binds with cytochrome c according to a paper with title “Salsolinol, a catechol neurotoxin, induces oxidative modification of cytochrome c”.
For ME that’s quite interesting as a modified cytochrome c would alter (likely hinder a lot) the Electron Transport Chain and with it the mitochondria and ATP production. The paper also says it releases iron from cytochrome c. That would both destroy cytochrome c and create massive ROS…
i had ME but I had/have NO PAIN. Sometimes I had some neuro pain in my legs I remember but it was not the main feature of my illness.
You’re not alone. A subset of patients have no pain at all – they’re weak and fatigued but without pain. If ME/CFS and FM are on a spectrum they’re on the far ME side.
It’s the opposite for me – the more I overdo it the more pain I’m in. On the other hand I can’t exercise like many people with FM can. I think I’m something like 60% ME/CFS and 40% FM.
At some point someone will get to the heart of these subsets.
Have you tried Thyroxine and T3 to see if you are just hypothyroid? AUnt Tammy said she has taken lots of T3 but is still ill with ME/CFS. Other people have responded to it they say and improved. Perhaps it does not matter how much T3 you take if it can not be absorbed into the cells? Dr Skinner said the TSH test is rubbish. You have to do all the whole panel of thyroid tests to see if you are hypothyroid and even then it might not tell you properly. OTHEwise, what is ME? Is there actually any evidence of encephalitis? or myelitis? No body gets these kinds of investigations so how can they say you have encephalomyelitis unless they can show you it on a SPECT scan? Has Aunt Tammy seen her thyroid panel of tests since she says she takes T3 a lot but still has ME. It happened to me too. I had symptoms and took t4 and T3 but I did not see any imporvement in symptoms. But why did my ME just go away suddenly? If you have a brain dysfunction how can it suddenly all disappear with no treatment? Isn’t it just that the thyroid kicks back in again and starts working?
HI – I come from the hEDS/POTS/MCAS/Fibro side of things.
I have been trying PEA for neuropathic pain (as recommended in this paper on MCAS pain, for fibro too).
https://tinyurl.com/y7kbw8d5
I’ve already been addressing dietary changes with great improvements. I now systematically wanted to try different supplements , and started with the PEA.
and tfu, tfu, tfu /knock on wood – I’m experiencing relief with my nerve problems – the tingling, the phantom cavity (in a tooth that no longer has a nerve…) and other mouth pains/sensitivities, the pain from the half-dead ulnar and median nerves.
I went back to reading the literature on PEA and it mentions that mechanism of action is through glial cells and neuroinflammation. And so I’m wondering if it could help with this aspect of CFS -?
Has anyone tried it here, with results?
(There are some studies on its use in fibromyalgia pain. From what I recall, is that there is some success when used with other modalities – allopathic medicines)
Interesting! Given my dental problems I should check this out.
What dietary changes did you make?
For more on PEA check out a Health Rising blog – https://www.healthrising.org/blog/2014/09/19/palmitoylethanolamide-pea-medical-food-fibromyalgia-chronic-fatigue-syndrome-mecfs/
What kind of dental problems do you have?
There are quite a few in EDS – do you have EDS?
(crumbling teeth, cavities out of nowhere, cracks out of nowhere, sensitive gums, gums that tear easily, etc)
The PEA seems to be helping with the nerve kind of pain. The permanent sensation of having a few cavities where there are none .
I did the usual. 12 years ago, I cut out wheat
– ah, I recalled now, you have mentioned your flavor of ME/CFS comes with pain, yes? –
and dairy, per a naturopath’s suggestions. We had no idea of any of my diagnosis back then. What I understood was that I was not allergic/intolerant to them necessarily . More like in the long run, their effects run my health down. I would sort of try a piece here and there and quickly it would enter my diet again. Then I’d stop. Wash rinse repeat.
What it helped me manage most directly and obviously has been asthma.
Last fall, I was eating a tiny pastry with cheese and had an anaphylactic reaction. I think it was quite clear this is problematic for me…
So I stopped completely since.
Later I found that article on treatment of pain in MCAS> It mentions that a subset of fibromyalgia patients are later found to have MCAS. So their pain responds well to addressing MCAS. They found eliminating wheat/dairy beef/yeast as the first line of treatment. [which, by the way, this is similar to the diet in some homocystinurias/methylation/remethylation disorders. i.e. MTFHR mutations…] Then antihistamines, PEA, and mast cell stabilizers.
Many hEDS people don’t metabolize medicines well, so I rather stay off those and stabilize mast cells other ways.
Here is that article, There’s a section on Fibro https://tinyurl.com/y7kbw8d5
What kinds of pains do you experience?
I have an all-you-can-eat buffet spread of them, and I have found some ways to address them.
That’s something that, I wonder:
I see that there is a lot of talk about neuroinflammation.
My question is: couldn’t’ wonky mast cells be a driver of that?
(at least in those of us with wonky mast cells)
I guess because I knew of MCAS before knowing about these theories/brain imaging studies. When I read about hyperalgesia in Fibro, I thought: they just described the problem with wonky mast cells. Could it be that is the mechanism behind it>?
@meirav, yes mast cell issues sure can cause pain. And lots of it. When mast cells degranulate and dump what’s in them, they cause all sorts of inflammation. There is however a connection with histamine and acetaldehyde. It can help clean it up and also ammonia. But we don’t want too much histamine either. Getting the receptors to work right and turn on and turn off when appropriate should be the goal. As histamine will actually HELP the immune system to activate a needed part of it. So blocking all histamine, may NOT be the best thing to do. See the thread Bayard has on the forum. (There are however other circumstances (COVID) where the mast cells when over activated, have to be stopped, in a hurry, and blockers may be the best option.)
Interestingly, I found out the hard way what too much fructose can do. We were drinking smoothies with protein powders and our cholesterol and triglycerides went up. It was from the fructose in the smoothies. And instead of getting healthy, we felt a whole lot worse. Now if I do anything too sweet, even a small amount, I pay for it. And because it can cause a type of addiction or craving, we tend to crave what we dont need. It helps boost dopamine.
Cutting out all fruits, grains and most nightshades and dairy has made improvements. And within 3 months cholesterol and triglycerides were back in normal range. Even white potatoes can cause a spike in many people blood sugar.
I think, to answer your question, can mast cells cause all this pain and issues…..it can most definitely contribute. But there are more pieces to the puzzle and they are all interconnected. We do believe that acetaldehyde is a big player too. Issues with dopamine and oxytocin, and also hypoxia and issues with blood flow. There are several things that need addressing. It is not just one thing.
And I’m as you are, and don’t tolerate medicines well either. So, supplements it is. And diet change. AND, pacing and not pushing beyond the halfway mark. Dejurgen calls it “leaving the party at the height of the fun”. Then we don’t go past our limits and don’t use all our reserves. But we reach our joy and have those memories to hold. Some of us have a hard time learning to do this and to stop ourselves when we feel our very best and not over do. (I know I do. And I have him to remind me. But I still crash myself.)
I did not try cysteine as other things point to that possibly not being a good choice for me. But I am using Yucca and DHM, bee propolis /pollen/royal jelly and a few other things. These are for acetaldehyde and ammonia. They seem to be making a difference. The other things, I need to observe a bit longer.
Acetaldehyde can also be caused from pathogens and that may need to be addressed.
I have talked about what I’m doing for MCAS on Bayard thread with histamine cream and nettle tea.
We will slowly speak of those other things. But we want to be able to explain our choices and WHY and not just throw a list of things out there with no science showing our reason for those choices.
@AcH Influenced, don’t rule out MCAS just yet. As it does seem it is connected to acetaldehyde. And the neuropathy seems connected. With my most recent added supplement, my neuropathy seems to be improving. And the red skin you speak of can also be associated with mast cell degranulatuon too. But it very well could be histamine trying to clean up acetaldehyde. So there can be both. And then there is ammonia released from all that. So we very possibly are a bit of a toxic soup going on internally.
And yes, I am sure dejurgen will like the link you posted. I sure did, and find it has a lot of very good information. Including connecting dopamine to this issue.
Oh, I also want to note…..there can be lack of a gene that causes one to not be able to break down alcohol and sugars properly called ALDH. It is mostly found in Asian decent. But I was able to look up the predominantly checked snps to see if mine were mutated and they are not. And I’m not of Asian decent either. But I do have other snps in that line that are mutated. So possibly there are other genes connected to that, that is giving issues……yet not yet recognized. Or there is just some wonky thing going on in some of our bodies making sugars and carbs turn into acetaldehyde and us not detoxing them properly.
Yes, I use it. There is one with Lutolin in it that gives more energy. I use the one without it at night. It works on the cannabidiol receptors. It also helps MCAS. It does increase its help over time. It can take several weeks for some to notice a difference.
I did find that it will, with time cause more brain fog. At least, it does with me. I don’t use it often because of that.
Here is a thread where I talked about it.
https://www.healthrising.org/forums/threads/potential-linking-fm-mast-cells-sleep-deprivation-food-intolerance-exercise-intolerance-and-me.6217/page-2#post-36652
Hi – I read the comments in the thread.
Have you tried n-acetylcysteine?
So many thoughts…
Is is easier to contact you through a post in the forums?
@Meirav, yes for me it is easier to contact that way. I get noticed when I have a message from there. We don’t get messages from the blog and they can get lost along the way as the next blog comes up.
Issie
Meirav and Cort : I have had strong mouth pain / rawness / sensitivity and gingivitis / gum receding problems. And ALL the nerves of my teeth hurt ( mostly the molars). This nerve sensitivity in my teeth drove me nuts sometimes and I started to clench my teeth because of it. Not the other way around ! you start doing that automatically because of the nerve pain sensations and you want to “clench that away”.
This was in the time when I also had mild polyneuropathy mostly legs (confirmed by EMG). I have had that strange combination of numbness AND pain. I have had tingling sensations in my fingers and when I tapped them on a surface I had that feeling as if I had been working with a jack-hammer or vibrating tool for too long.
Also the painful throat thing and I had strong lymph-node problems sides of my neck and upper chest.
Now here is the interesting thing : as I made my disease less severe all these problems got less over time or disappeared. No more tooth nerve pain / polyneuropathy and such. Much less mouth rawness / pain and gum problems. But when I start eating badly again with lots of carbs and such the mouth rawness / pain and gum problems and all the rest get immediately worse. And neck lymph-node problems get noticeable. I can make these things better or worse at will somewhat !
In my case they are all related and I know that the acetaldehyde is causing that because I have my own “acetaldehyde-o-meter” that I can see in the severity of my red face and face-flushing feelings and redness on my upper arms (coupled with more brain problems/ -fog and all the others symptoms that get worse). This gets all more then. My explanation for it all is that the acetaldehyde has an effect on the nerves ( causing pain AND numbness) but acetaldehyde is also a strong irritant and it irritates the mouth / throat areas on a living tissue level from the INSIDE out. And it irritates the lymph nodes so they start producing fluid.
> Meirav you mentioned the n-acetylcysteine I tried that myself to see if it would make a difference with my acetaldehyde problem but it did nothing for me and neither did similar supplements for that purpose.
I too had face flushing, the skin on my knees and one thigh went numb and I had a peculiar feeling of a tingling, chilly wave through my body from my feet upwards and diagonally… They’ve all gone now.
I do notice my pain sensations go down when I’m doing better. My dental problems just include numerous cavities and now crowns and root canals plus at times my teeth just really ache – all of them…Really weird.
The teeth, mouth, phatom nerve pain( tooth gone) is driving me nuts and just keeps getting worse for me over the past two years or so… No dietary changes even put a dent in it. It’s the worst… Maybe I’ll try this pea stuff.. don’t need anymore brain fog though!
Yes! My Dr. wanted to say that I was causing my teeth pain from clenching, when it was the other way around, that I was clenching FROM the pain! Needless to say she didn’t quite get it!
I hate to being this up, but you guys with teeth pain and root canals and caps…….look into cavitations. It could be infection set in at the bone and will have to either be cleaned out and/or ozone put into the cavity/socket area hoping it will get the inflammation out. Or both! It can go on for a very long time and until addressed, you may not get better.
There are two other things I know affect my tooth ache:
* Enlarged sines; apparently we have sines on top of the top row of teeth too and they can press against the nerves of those teeth. That pain can radiate as if all teeth hurt.
* The jaw bones can be painful, again radiating to the teeth. Immune reactions get my bones aching easily. To my surprise, bones have mechanisms to actually sense pain. They are more then solid calcium deposits. Just as AcH Influenced, I have “acetaldehyde flushed” areas below my eyes, with visual damaged capillaries as if I had an alcohol problem.
Like Issie, I think the red and glowing effect is due to a modest but frequent mast cell activation over there. That would be an immune reaction very close to the jaw bones. There do exist receptors for acetaldehyde too, so this could point to an acetaldehyde induced mast cell response.
https://www.npr.org/sections/health-shots/2020/05/27/862963172/how-the-lost-art-of-breathing-can-impact-sleep-and-resilience
Some very helpful information which this scientific journalist learned about breathing, with resulting changes in neurological or endocrine balances, health and longevity.
Here is an interview of the guy that wrote the book on “Breathe”. I enjoyed watching it. And I’m learning from the guy who wrote “The Oxygen Advantage ” too.
Changing how we breathe can make a big difference.
https://youtu.be/wl4J2LMXcx8
I’m working on absorbing this information. I’m very intrigued. I’m wondering if anybody else benefits from molybdenum/molybdate?
I’m wondering if anyone else has low blood Uric acid levels? Anybody else benefit from lipothiamine?
I have a permanent tan/red flush on my chest. Developed alcohol intolerance ten years ago. Gall bladder removed eight years ago. I’m not diagnosed with ME/CFS, but I am diagnosed with autoimmune issues, EDs, migraines, MCAS etc..
Hi Robin,
I have (only) normal uric acid levels despite being family predisposed to have good chances for high uric acid.
Uric acid is an anti-oxidant so if you have plenty of oxidative stress some may react away in the blood.
Also, uric acid is hard to keep in solution. If you have for example below average cold limbs it can easily crystallize and cause inflammation around the joints even if you have low uric acid. Also having acidic blood can cause it to crystallize at lower then expected levels.
Once crystallized it goes not easy into solution again. Other (scar like) tissue can encapsulate it or the immune system can try to remove it by for example bombarding it with oxidative stress. As uric acid is an anti-oxidant that can remove it although it likely requires plenty of oxidative stress. If you regularly have joint inflammation some while after “health issues” this mechanism may be part of it.
Just high spikes in blood lactic acid are IMO enough to seed a new round of crystallization. Big spikes in blood lactic acid can occur for example after increased exercising.
My test to determine if it is LIKELY, not for sure, uric acid is the warm socks test.
It goes as follows: You have two really warm pair of socks. Both must however not be tight so that they are not pressure socks.
In one situation you wear the short but warm socks that exert no pressure on your legs. On top of it, you wear loose trousers that do not isolate well. Notice where your legs are inflamed.
In a similar situation you wear the long and warm socks that exert no pressure on your legs. On top of it, you wear loose trousers that do not isolate well. Notice where your legs are inflamed.
If you have got inflammatory pain just on top of your socks each time (make sure the socks’ elastic doesn’t cut into your legs) and it repeats itself, you have IMO a good chance that uric acid starts to crystallize on the onset of the the colder zone of your legs.
By wearing different socks in identical situations the inflammation follows the cold zone. Of coarse it doesn’t follow it by the minute. Each experiment takes hours to a day. The only requirement for the experiment is that the problem is bad enough for it to no only deposit on your joints but also on your legs.
If that is not the case, you can for example make some “elbow tubes” that keep that place warm and see if inflammation disappears (and appears somewhere else).
During hot weather this test obviously works less good.
Dejurgen—absolutely fascinating. Thanks for helping me learn more about uric acid. I went to a rheumatologist for the first time about a year ago because I was interested to learn if I was HLA-B27+. Based on my exome data, snp data and clinical issues (degenerated c-spine) I expected to be. I am. She tested me for uric acid and told me I was one of the lowest she had measured. But had no insight for me about its implications. I told her I was concerned it had implications for neuroinflammation—my symptoms always present similarly to MS. I’ve subsequently studied my genetics on uric acid and I’ve found I’m at least genetically wired to have low uric acid. And with my tendency toward oxidative stress I’m probably using up the little bit of uric acid I make.
Issie—I’m curious to learn what everyone uses successfully to help this acetaldehyde issue. I’ve tried DHM but it caused me pain. I do well with an N-AC with molybdate in it. I do well with lipothiamine and magnesium taurate. There is a new TTFD thiamine on the market I may try as well. i will need to trial some Yucca.
Ach—my skin is like your skin. I’ll be curious to learn more about your self-care. What has helped you make or metabolize acetaldehyde? Are you also a slow acetylator? Have you studied your genetic underpinnings?
Martin-Hashimotos was my first diagnosis back in the 90s. I’m on a t4-t3 medication and it hasn’t resolved my fatigue symptoms but it has reduced my fasting glucose and dry eye symptoms.
Cort—thank you for this excellent site. I admire all that you do.
@Robin. I found I have to use Yucca with DHM. Seems to be a connection with the detox of acetaldehyde and ammonia. Otherwise DHM made me feel worse too. And not all DHM worked the same. I tried different brands and found one better than another.
I use the traditional form of B1 and find it worked better for me than benfotiamine. Though it seems more talk of benfotiamine and benefit when it comes to ME/CFS and POTS. But I am not finding that with myself. And have tried both.
There also seems to be some connection with some using P5P the activated form of B6. But I found that gave me vivid dreams, restless sleep and what Dejurgen and I are calling “narcolepsy light”. It is suggested to use with zinc. But I could not take the type of sleep it gave. I’m super sensitive to things and even a tiny amount was too much. Though it did seem to help neuropathy. But one of the new things I’m doing is helping neuropathy better and having better sleep and less issues. Seems to be more beneficial with less side effects. And should improve the detox of these things and help oxygenation. Will talk about it later, when I get it better sorted and effects and affects better known.
@Robin, forgot to comment on you being positive on HLA – B27, my dad had akylosing spondylitis too. When checked in my early 40s they said I have signs of issues in my sacrum that was starting. Back then genetic testing was really expense and they didn’t do it as easily or often. They did a lot with MRI and symptoms. I’m not as rigid as my dad was, and I don’t feel it is developing. Hoping all my preventative things are helping that.
I do however have EDS (that was confirmed by same doc) and that has caused spinal stenosis with lots of calcification. But my pain is much better now, with what I’ve been doing in the last year.
Inflammation is a huge factor, genetics a big player and faulty autoimmune response.
@Robin, I do take molybdenum occasionally. The other things I use seem to work pretty well for removal of ammonia. But when I need additional help, I use molybdenum. It is however a metal and I do try to be careful with it.
Also I use Thiamine. I am finding that really helpful. It has helped in more than one way. It has not only been found to help ME/CFS people but POTS people too.
I’m looking forward to reading what you sort through. Very interesting to read about using the Yucca with the DHM. I, too, am extremely medication/supplement sensitive. I’ve spent time learning about porphyria, CYP450 issues, BH4 recycling and methylation. In addition to EDs genetics, B12 metabolism, Beriberi and dysautonomia (ansar testing indicates I experience both sympathetic and parasympathetic withdraw). Now it’s time to learn more about acetylation and acetaldehyde.
@Robin, I have issues with P450 pathways too. The liver detox issues are huge. I’ve known this issue since in my 20s. A lot of what we are discovering, explain a good bit of this. The things you speak of are also things I have looked into and/or have issues with myself.
I did not find however, the treatment suggestions for pyroluria to be something I could do. I already discussed P5P and the vivid dreams that caused. But issues with the Kyrenine Pathway does seem to fit with problems with glutamate and the issues with the sympathetic nervous system.
There is another thing I use to take ammonia and acetaldehyde down, it is a combo product of bee propolis/bee pollen and royal jelly. I sometimes interchange it with Yucca. It also is a good help for MCAS issues. I do have a CBS mutation. (Issues with sulfur pathways, and that forms more ammonia.)
NOTE: I only say what I use and am not suggesting anyone do this. Just saying a few things that are working for me. Make sure you do your research before you take supplements. They are as powerful, or more so, than medicines.
@Robin, neither molybdenum nor other supplements I tried for my own problem to “scavenge” the acetaldehyde or are supposed to make the processes that get rid of acetaldehyde work better did anything for me. I have been red from head to toe when I was really ill but these days apart from a red face I had / still have indeed a red complexion over my chest, a sort of reddish mini-blotchiness effect.
An interesting thing btw is that if I got a sun tan at exposed areas especially upper chest with that redness part when wearing a v-collar shirt or something like it that that tan stayed for months longer than usual ! I also felt the sun giving much more uncomfortable feelings of “burning” on my skin.
When I was really ill I often had a lingering headache though not extreme but I was VERY worried that it would develop into a migraine but that luckily never happened. I am not saying that this is all the case with you but I believe this particular problem brings out a persons own personal weaknesses like vague auto immune issues and maybe possibly MCAS related things ? Who knows what more ? A large part of this acetaldehyde problem is that acetaldehyde is an irritant I think, the body does not like it and it also gives itchiness and eczema and such. When I was at my most ill I was very worried I would develop an itchy skin disease….
Yep it gives you alcohol intolerance or at least the whole experience is different and not as it used to be.
@Robin I had quite severe dry eyes and mouth. Waking up in the middle of the night and I had to use eye drops and many times during the day. I first thought I had Sjogren syndrome because all the other symptoms and rheumatic things also looked like an auto-immune disease. The dry eyes / mouth slowly went away as I made myself better.
I don’t believe genetics are involved with this problem more a general weakness in some people so they can develop this problem or after an illness. I can understand that you want to know how I made my symptoms better but seriously I just want to avoid the topic at the moment because it is such a big one and will lead to so many questions I just can’t bring up the little energy I have to do it…. One day I will have to tackle the subject but in the mean time just follow the acetaldehyde topic / my website I hope you understand… But in general low-carb diet + all sorts of natural anti microbial stuff but it is an enormous struggle to get rid of this problem been busy trying it for years now.
It’s just good to read that you’ve made progress. I’ll look forward to what you’ll share when you are able. I tend to look at genetics because I’ve had some success in making some improvements to my health by understanding my genetic variations (I.e., my many medication/supplement reactions can be accounted for by my CYP450 variations).
Your thoughts on genetics made me check some of my snp data on ALDH2 gene. I do have some homozygous variants on that gene.
I also found this 2019 article on endothelial function and the ALDH2 gene: https://res.mdpi.com/d_attachment/biomedicines/biomedicines-08-00004/article_deploy/biomedicines-08-00004.pdf
Lots for me to digest here.
@Robin, nice article. It shows the widespread dysfunction that our genes can have and consequences of them not working properly. Few have the ability to figure out their genetic dysfunction or pay close enough attention to their “symptoms” to get a clue what to associate their dysfunction to. But when we start to sort it, it’s an amazing, exciting discovery.
Dejurgen and I have so many links to support what we have been researching and connecting for about a year now. We also have been doing self experiment to try to correct these dysfunctions, and are making improvements, finding a lot of WHYS and obtaining some “purple bandaids”.
It is a lot to take in, a lot to sort, and a lot of connected pathways that may need addressing. But it all makes perfect sense and we feel it will be HUGE!
Dejurgen has done a couple of recent post, starting to put our research out. We are still in self experiment to try to get these pathways tweaked and proper support sorted. We are getting a lot closer.
As mentioned, we are all different. What he can take, supplement wise, I can not. (My genetics are really wonky. But we are finding ways to work around that.) We will put more info out as we get the science sorted and the tweaks a little more consistent. It is not an easy thing to manage. And for sure can affect so many things in the body and possibly have many different illnesses connected.
Thanks for that link. I enjoyed reading it!
Issie
Just read about Jen Brea. DId not realise she recovered. But everyone missed something. She had thyroid surgery for cancer. So she was just hypothyroid and that is it. Thanks
Lol come on with that simplification. She still had symptoms after thyroid surgery in fact she got worse. She recovered after CCI surgery.
Nah. She WILL have symptons after thyroid surgery if she is not medicated enough. This is what happened. Nothing to do with CCI surgery which is a misnomer. Keep taking the chocolate though f it helps you feel better. Thanks
Hi!
I’ve been reading comments and forums.
I’m a newbie, you guys are way ahead into things.
I want to thank you. Reading through the forums
Dejurgen mentioned that amino acids tend to be low in ME.
I looked around the internet for published studies and there they were.
I ran tests for amino acids about two months ago, when I was crashing badly.
It was peculiar because most of the values
were towards the left side of things – almost borderline low / borderline / low
EXCEPT for homocysteine, methionine, and cystine. Those were smack center of the range.
If I look at it in terms of patterns – they are definitely higher than the rest.
I wonder if if could be possible that testing when I was crashing masked a hyperhomocysteinemia?
I have symptoms, and I respond to the corresponding supplementation.
The metabolic disorder doctor wast not convinced…
It’s a good thing there are direct to consumer labs now – are there recommendations for how to go on about this, in the least expensive way?
I realized too that I have done more nutritional changes thru the years. Some not intentional.
I didn’t have any cavities until my late 20s,
and then I had 4 or 5 at the same time. Nothing in my life had changed to precipitate them. I kept having teeth problems.
At some point I cut out processed sugar, for other reasons. After about two years of this, I switched back to my old dentist. Looking at my xrays, he specifically asked me if I had cut out sugar. How did he notice. He said the enamel was healthier and stronger.
Same thing is happening to my nephew…out of nowhere, tons of cavities at once. high glucose levels.
I also had peculiar weight gain. I could put on kilos really quickly and I was not changing the way I ate. It was very hard to lose them. Until suddenly I would drop them really fast. I realized it was wheat. My body responds this way to it. Add toast for breakfast every morning, and surely it shoots up. Except, except… one summer I lived in taiji village – Taiji all day, 6 days a week. I ate wheat, eggplants (allergic) . Yet, no allergic reactions. And my weight went down. And my skin was calm. And so was my heart and the POTS symptoms. That was also where it was first pointed out I had soft joints, by the teachers. No doctor had ever noticed the hypermobility…
I tend to be allergic to a lot of fruits, so that went out too.
This way I have been able to keep my blood glucose levels from creeping up.
I address MCAS by avoiding triggers, many revolve around food. I also got rid all hard cleaners, and stopped using fabric softener, etc. I brought my reactivity levels way down. I also drink nettle infusions – brewed overnight. I first started 13 years ago to use it during hay fever season. Now that I learned about MCAS, and quercetin, and blood irregularities – pre-industrial medicine times, I read it was regarded as a ‘blood tonic’, I read also is rich in B6, which I may need – I figured to try it daily. I feel it does something.
I then added the PEA and this seems to be addressing nerves. I hope also stabilizing mast cells.
Issie – which kind do you use? The powder form I use, has a type of lentil powder. This is protein and if one has homocystinuria CBS gene, could that cause brain fog? I talked to a woman with classical hmctnria – she calls it protein fog.
I kind of figured, that anti-histamines just block the action, and I also read you can get rebound effect once the effect wears off.
I feel the same way in regards to medication for my heart rate with POTS. If the theory is my heart is pumping like crazy to get blood flowing back up again, would taking a medicine that would not let it pump like crazy mean my blood would pool even more in my lower extremities? Maybe this is not what is happening, at all.
:AcH – the symptoms you describe are quite familiar to me and your measuring meter… I used to refer to them as my body being a live-wire, sensitive and raw and touchy. I later learned about MCAS, so I work my way backwards to figure it out- what could it have been that I’m reacting to?
I have found that curcuma lessens my facial skin redness, irritability, rawness. Both as a supplement AND/or topically. Calendula in whatever form, even steam from dried flowers. Avene has a line for ultra-sensitive and reactive skin. It not only hydrates without causing reactions – it actually stabilizes my skin for me. I have no idea which ingredient does this. You can even bypass using tap water to wash your face (if it irritates you) with their spray water. My skin is very very dry as well.
Ah – I have one very specific symptom. Hair shedding. Dr. Afrin has it in his literature as an MCAS symptom. It doesn’t seem connected to other reaction symptoms in my body…
These supplements I have been trying along the homocystinurias – they directly work on that. The NAC, methylfolate, and B6 thus far. I’m not sure if it is sustained. I have not started taking them on a daily basis yet.
: AcH
the NAC works on my releasing muscle stiffness and spams. And the hair. I take it in liquid form. what did you want to address when taking it?
By the way, I’m gathering Dr Afrin treats MCAS by addressing methylation as well.
“I wonder if if could be possible that testing when I was crashing masked a hyperhomocysteinemia?”
If you crash “often enough” it may actually never get to the point of being too high. If so, IMO technically it wouldn’t be hyperhomocysteinemia as that requires homocysteine to be too high.
I don’t know however how rapid it would pile up to too high and if that period is longer or shorter then the period between crashing. You even might not need real crashes to have all amino acids lowered compared to if you would not have ME. With me, each night acts like a mini crash as to speak.
It seems there is some “challenge” in your CBS pathway or one closely related to that and as that is connected to several others those might share part of the “challenge”. But at the moment seeing how this complex interaction works in practice is too difficult for me. Practice is often more difficult then theory.
When you’d ever get the luck of improving enough you might run into issues with homocysteine. That could be interesting to keep in mind.
Watching out if sulfur rich food triggers MCAS may be smart too. And looking out for episodes of high blood ammonia (and IMO related night sweats when crashing) could offer other clues.
What product of Avene do you use? I’m interested to see what can stabilize this effect (that I have too) as it could point to how to fight back more problems.
This cream and cleanser
https://www.avene.co.uk/face/specific-care/hypersensitive-allergic-or-intolerant-skin/skin-recovery-cream
https://www.avene.co.uk/face/specific-care/hypersensitive-allergic-or-intolerant-skin/extremely-gentle-cleanser-lotion
For the past 13 years, there is a strange odor in my room. It’s from my sweat, what lingers behind on bedsheets and clothing. I have to constantly wash them. Sometimes it is so bad, it reaches out into the hallway.
It’s noticeable to others. No one can describe it. I think it’s simply foul.
Recently, others can even smell it on me, when I got back home from being outside. It waxes and wanes.
There was a dead mouse in the shed, and something about the smell reminded me of the smell in my room. I looked up what is the substance responsible: sulphur, methane from the body decomposing (the mouse’s, not mine)
Started noticing it only in the last 13 years.
I crashed very badly towards the end of April, when I had the testing done Days in bed, in darkness and quiet. I’m a bit better now – but worse than winter. PEM sets in much faster now.
I experience the night sweats. Also, the ammonia fumes escaping with my sweats. In fact, a couple of days ago I had a small chicken liver, and two-three hours later, I sweated a bit and the ammonia choked me a bit. Sometimes it stings my eyes. What could this be pointing to?
I looked up which food are sulfur-rich. There’s plenty of them I don’t eat because they make me sick. Could this be a symptom due to sibo as well?
After I saw the presentation on the metabolic trap and IDO enzymes by Robert Phair, I learned that chemical reactions represented on ‘paper’ are much more complex than A + B = C. So I look at the pathways in methionine synthesis and think: there is so much more information in this graph that we don’t see.
I.. was a fairly convinced I had a homocystinuria. Researched homocysteine and effects on the body. There are some very compelling studies and theories of how it affects muscles, and even the central nervous system. And then my test results came back normal for homocystinurias, and I thought: I know very very little. It was easy for me get carried away and build up theories – need ways to test to provide insight, directly or indirectly. And be able to work back and forth between the two. Need to remember that
If not a homocystinuria, what can be causing the foul odor AND that the nutrients work well for me?
I looked up homocysteine and acetaldehyde… I just quickly scanned the links, mentioned that it the second can affect the first and methylation as well. And acetaldehyde may also affect B6? Is this what you and Issie have been studying? I look forward to what you will both share and find works for you.
@Meirav, sounds like you have made some positive changes. I also have cut fruits, sugar and grains from my diet. I do better without them. And nightshades and lectins are an issue too.
I think some of us have issues with protein in our diet and how we break it down and assimilate the aminos from what we eat.
It may very well be the issues with methylation play a big role in it. I have a lot of issues there with those. But my trying to address it, has not gone well. Things that should send things in the right direction, backfire with me. So, figuring out how to work around that is a challenge.
The DHM I prefer is out of stock. I hope that it becomes available again. Thankfully I had a backup bottle. The one I chose I got from Amazon and is by Naturebell.
A lot of the supplements you are trying, if they work properly with you, may be very helpful. I had hoped to have a good response with them, but didn’t. I hope they keep working. Some find adding zinc important with the B6. Especially if issues with pyroluria. Connection to Kyrenine pathway.
There is a lot written on mast cell here. I have it very bad and am trying to reset receptors. Slow process. But off all the antihistamines. Making improvements. Look for a thread on the forum by Bayard.
And yes, hair issues can be a problem. Nettle tea also increases histamine initially and then has a very calming effect. I drink it an hour before bed and that improves sleep.
Dejurgen mentioned issues with sulfur and CBS. Keep a watch on the NAC in that regard.
The PEA I use is from the Netherlands. One has lutolin in it and is good for day time as it gives energy and helps with MCAS. The other is plain, also from the Netherlands. I don’t use often now as they did seem to increase brain fog. But I still do occasionally use them.
Good for you, getting chemicals out of your life. As clean as you can have your surroundings and what you use on your body, the better. I use white vinegar as a clothes softener and use only baking soda and a little borax as my clothes detergent. Works beautifully. And not toxic.
Sounds you are on a positive track. Keep going!
Issie –
I’m cautious, because when I first tried the methylfolate, NAC and B12 – it was only for a few days. About a month later and a half later, I crashed very badly – perhaps the worst ever? I had also stopped the nettle infusions. The seasons were changing too, and the heat exacerbates the hyperPOTS. Not having access to pool/acupuncture and losing muscle tone during corona – I lost all my muscle tone.
I’ve gotten a bit better since but I’m experiencing more frequent crashes, PEM sets in much quicker.
I do take the methyfolate more often now, when the muscle spasms are too much, it’s a little bit hard to resist not doing so. Not sure if this is a good idea or not.
It’s tricky.
MCAS and nerves are better.
I’ll check out DHM.
I look forward to what you and Dejurgen share 🙂
DHM addresses acetaldehyde, but I have to fake either Yucca or Bee propolis/Pollen/Jelly with it for ammonia. If I don’t it makes me naseauous.
There are 2 new things I’m trialing and they seem to be the added boost I needed. Need longer to observe.